Parkinson's Disease is the second most common neurodegenerative disorder. In Germany alone, almost half a million people are affected. The focus of the disease is the progressive degeneration of dopamine-producing nerve cells in a certain region of the midbrain, the substantia nigra. Misfolded proteins are the cause. Until recently, it was unclear why damage is confined to specific nerve cells. A team or researchers led by Frankfurt neurophysiologists has now defined how this selective disease process begins using a genetic mouse model of Parkinson;s disease.
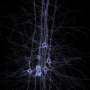
The progressive death of a certain type of nerve cells – dopaminergic neurons - in the substantia nigra causes dopamine deficiency, which is the major cause for the motor deficits in Parkinson patients. Although it is possible to therapeutically compensate the dopamine deficiency for a certain period of time, by e.g. administration of L-dopa or dopamine agonists, these therapies do not stop the progressive death of neurons.
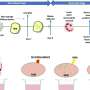
In the last two decades, researchers have identified gene mutations and toxic protein aggregates to cause neurodegeneration, with the protein a-synuclein having an essential role. Until recently, it was unclear why only specific types of nerve cells, such as dopaninergic neurons in the substantia nigra, are affected by this process, while others, also expressing the mutant a-syncuclein such as dopaminergic neurons in the immediate vicinity, survive the disease process with little damage.
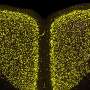
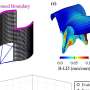
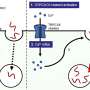
The research group led by Dr. Mahalakshmi Subramaniam and Prof. Jochen Roeper at the Institute for Neurophysiology at the Goethe University, in collaboration with researchers from Frankfurt's Experimental Neurology Group and from Freiburg University, demonstrated for the first time how sensitive dopaminergic substantia nigra neurons functionally respond to toxic proteins in a genetic mouse model. A mutated a-synculein gene (A53T), which causes Parkinson's Disease in humans, is expressed in the mouse model.
In the current issue of the Journal of Neuroscience, the researchers report that the sensitive dopaminergic substantia nigra neurons respond to the accumulation of toxic protein by significantly increasing the electric activity in the affected midbrain regions. In contrast, the less sensitive, neighboring dopaminergic neurons were not affected in their activity. "This process begins as early as one year before the first deficits appear in the dopamine system, and as such it presents an early functional biomarker that may have future potential for preclinical detection of impending Parkinson's Disease in humans," explains Prof. Jochen Roeper. "The potential for early preclinical detection of subjects at risk is essential for the development of neuroprotective therapies."
The Frankfurt group, also identified a regulatory protein, an ion channel, which is causes the increase in electric activity and the associated stress in nerve cells in response to oxidative damage. This channel provides a direct new target protein for the neuroprotection of dopaminergic neurons. In brain slices, the dysfunction of this ion channel acting as an "electric brake" for dopamine neurons was reversible just by adding redox buffers. If therapeutic drugs could reduce the channel´s redox sensitivity in future mouse models, the death of dopaminergic neurons in the substantia nigra might be prevented. Currently, the researchers are studying whether similar processes occur with other Parkinson genes and in aging itself. "The long-term objective is to investigate the extent to which these results from mice might be transferred to humans," says Roeper.
Journal information: Journal of Neuroscience
Provided by Goethe University Frankfurt am Main