Scientists unveil powerful method to speed cancer drug discovery

For decades, researchers have struggled to translate basic scientific discoveries about cancer into therapeutics that effectively—and with minimal side effects—shrink a tumor.
One avenue that may hold great potential is the development of drugs that interfere with interactions between proteins, which are often disrupted during the formation and spread of cancer. Deciphering these interactions, however, has proven difficult and time consuming, leading to doubts about the practicality of this approach as a route to new therapies.
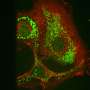
Now, Salk scientists have developed a highly sensitive, new method that enables them to detect fleeting protein interactions that play critical roles in the development of many diseases including cancer. The approach, published November 20 in Cell Reports, could dramatically accelerate the identification of many potential new drug targets and provide an immediate platform to screen for badly needed new drug candidates that disrupt abnormal protein interactions.
"The number of protein functions that are currently targeted by drugs is incredibly small compared to the total number of protein interactions that could be targeted for therapeutic benefit," says Geoffrey Wahl, a professor in Salk's Gene Expression Laboratory. "If we can crack the nut of screening for drugs that disrupt cancer-relevant protein interactions, this will be an enormous breakthrough and could have implications for many other fields as well."
Yao-Cheng Li, a staff scientist in Wahl's lab and first author of the new paper, explains that their method focuses on one of the two kinds of protein-protein interactions. "One type generates very stable protein complexes that remain together," he says. "But many other proteins display a touch-and-go kind of interaction—they bind, then fall apart. It's these latter interactions that have been the most difficult to detect."
To help visualize these brief, transient interactions, Li and Wahl turned to a molecule called luciferase, an enzyme that generates bioluminescence of the sort used by fireflies to make their bodies glow. The scientists adapted an old method in which luciferase is split in half to make two non-functional fragments. The scientists attached each half of the luciferase to two proteins of interest so that if the proteins associate for any period of time, luciferase's two halves are brought together and emit light. The secret to the new method comes in the many tweaks and improvements that Li added to the system, which is symbolized by the acronym he and Wahl apply to the method—ReBiL—which indicates "recombinase enhanced bi-molecular luciferase complementation."
"It works like a bulb and a lamp," says Wahl, who is also the holder of Salk's Daniel and Martina Lewis Chair. "Neither one lights up without the other. The ReBiL method provides a very fast and easy way of seeing whether the bulb will fit into the lamp socket."
To test the method, Wahl and Li applied it to the interaction between two proteins, Ube2t and FANCL, that's been notoriously hard to observe and had never been seen in living mammalian cells. These proteins are important because they are involved in the cell's ability to detect and repair DNA damage, a function that is often disrupted in diseases. Mutations in FANCL, for instance, cause rare blood disorders and dispose people to cancer. The ability of ReBiL to reveal the stealthy FANCL-Ube2t reaction suggested the method could be a powerful technique for observing other similarly challenging interactions.
The Salk scientists then used ReBiL to study a promising target for cancer, the interaction between the proteins p53 and Mdm2. The function of p53 is affected in almost all cancers and, in many cancers, too much Mdm2 prevents p53 from functioning properly. Hence, a major goal of cancer scientists has been to develop drugs that prevent Mdm2 from binding to p53, and to thereby activate p53 to kill the tumor cell.
Wahl, Li and their colleagues used ReBiL to confirm that some drugs work as expected to prevent Mdm2 from binding to p53. On the other hand, when they applied their method to a new class of promising drugs called stapled peptides, they found that the drugs had difficulty entering cells and had the unexpected and unintended ability to kill cells by punching holes in their protective covering (the membrane). Despite spending millions of dollars to develop these drugs, this dangerous side effect was not observed because previous methods did not reveal it. ReBiL provides a fast and simple way to try to improve stapled peptides to enable them to get into the cell, bind to their targets and kill cells by the specific route they were designed to use.
The fact that ReBiL can be used to study living cells (as opposed to many older methods that use isolated proteins from cells to determine their interactions) makes it an ideal way to observe these unexpected side effects and to modify the drugs to eliminate them, Wahl says.
"We think the method is already so good and so versatile that we're already applying it to many different questions," Wahl says. "It has applications from understanding many growth regulatory pathways and for understanding critical processes that should lead to the identification of the targets needed for the development of new therapies. Not unexpectedly, academics in many fields as well as companies have already shown interest."
Wahl and Li imagine ReBiL being used in the future to discover new interactions between proteins that might serve as cancer drug targets, as well as being used in robotic systems to identify drugs that disrupt protein interactions. They also foresee using the technology to help avoid the off-target effects they have already identified for stapled peptides.
Alan Saghatelian, a professor in Salk's Clayton Foundations Laboratories for Peptide Biology, says ReBil is an important new technological platform for scientists. "This will have a powerful impact on the development of new medicines and the discovery of novel biological mechanisms," says Saghatelian, who was not involved in the study. "The results demonstrate the value of this approach in prioritizing drug candidates and understanding mechanisms of drug action."