The drugs were designed to keep cancer cells at bay by preventing their growth, survival and spread. Yet, after clinical trials, they left scientists scratching their heads and drug developers watching their investments succumb to cancer's latest triumph.
The drugs worked like they were supposed to, shutting down the cellular receiving dock, EGFR (for epidermal growth factor receptor), on which many cancers thrive, but still the drugs failed to stop cancer in most patients.
It was Xiaojun Tan, a graduate student in Richard A. Anderson's lab at the University of Wisconsin-Madison, who began to crack the case after an unexpected observation he made while studying the locations inside cells where EGFR can be found. His subsequent investigation revealed how cancer was evading these drugs: by sneaking through the cellular back door.
The results of the study were published today (Jan. 15) in the journal Cell.
"What we see here is quite different," says Tan. "It's an alternative strategy to promote cancer cell survival."
Though the drugs were doing what they were shown to do in laboratory studies—inactivating EGFR—Tan found that cancer cells are also able to use the inactive form to thrive. The findings could have tremendous impact on human health and huge economic impact for drug developers, says Anderson, a UW-Madison professor of pharmacology.
In normal cells, EGFR functions to the benefit of the cell. But in a large number of cancers—ranging from more common ovarian and skin cancers to rarer, aggressive cancers like glioblastoma in the brain—the gene controlling EGFR is reprogrammed and too much of it gets made, leading to unchecked growth and cancer cell spread.
"Hundreds of thousands of patients every year have tumors addicted to EGFR," says Anderson. "It has implications for millions of cancer patients worldwide."
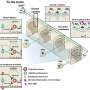
Tan and his UW-Madison co-authors, Narendra Thapa and Yue Sun, found that inactive EGFR is, unexpectedly, involved in a process called autophagy, which literally means self-eating.
Healthy cells use autophagy as a means of tightening the molecular waist belt during meager times, when resources are in short supply or when the cell is stressed. Essentially, the cell consumes its nonessential but energetically costly contents in a move to survive.
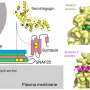
Cancerous cells also use it to survive stressful conditions, like they may experience when packed tightly into the middle of a tumor. Tan found that cancer cells with excess EGFR also have high levels of another protein, LAPTM4B, that helps escort the inactive EGFR to the place inside cells where autophagy begins.
Once there, the inactive EGFR can help set off a cascade of cellular changes that triggers autophagy to promote cancer cell survival.
It's the first time LAPTM4B has been implicated in initiating autophagy, Anderson says, though the study shows it's not required for EGFR to trigger the cascade if inactive EGFR finds another means of reaching the autophagy machinery.
The research team's findings suggest that, to stop cancer, the original drugs developed to inactivate EGFR could be combined with drugs that block autophagy to deliver a one-two punch that seals the front and back doors cancer has used in its sneakiness.
"We predict it could be an incredibly effective way of treating cancers," says Anderson, who notes both types of drugs already exist and have FDA approval. "It could have potentially a very rapid impact on cancer treatment."
Of Tan, Anderson says: "He found this while examining another cellular question; that's how basic science works ... I think his work will have tremendous impact on human health and I'm very proud of that."
Journal information: Cell
Provided by University of Wisconsin-Madison