Can we cure Huntington's disease?

I didn't cry until page 123 of Lisa Genova's terrific new novel Inside the O'Briens. That's when 44-year-old Boston police officer Joe O'Brien tells his four young adult offspring that his "weird temper"; his frequent toe-tapping, shoulder-shifting, and eyebrow lifting; and his inability to sequence the events in a routine crime report, are all due to Huntington's disease (HD).
As a boy, Joe believed the neighborhood talk that his institutionalized mother was an alcoholic. He remembers his skeletal, writhing, grimacing and grunting mother as a monster, not as someone suffering from a neurological disease only trying to say "I love you" to her terrified son.
But it is a passage on page 186 that inspired this post: a genetic counselor tries to positively spin the worst-case scenario to Joe's 21-year-old daughter Katie, who is agonizing over whether to learn her 50:50 genetic fate. If she has inherited the mutation, she likely would have at least 15 to 20 years before symptoms begin.
"A lot of things can change in that amount of time. There's plenty of real hope in the research being done. We could have a really effective treatment or cure by then," the counselor says.
I've said similar things to patients who have a genetic disease in the family. But a treatment or cure may be asking a lot—slowing or amelioration of symptoms might be what's actually possible. I inwardly cringe at highly-publicized efforts to "find a cure" by throwing money at a medical problem – from Richard Nixon's War on Cancer to today's Stand Up 2 Cancer—as if the answer has been hiding and we just haven't seen it. Those campaigns make me think of HD. Sometimes money isn't enough.
Slowing, not curing, is OK
The privately-funded CHDI Foundation has been donating $100 million annually to HD research for years, "to develop drugs that will slow the progression of Huntington's disease and provide meaningful clinical benefit to patients as quickly as possible." (Disclosure: I've written research reports for CHDI.) Like treatments for HIV infection and cancer, slowing disease progression and easing day-to-day life are valid goals, and may be enough to change a terminal illness into a chronic disease.
With those caveats, HD research may finally be turning a corner from preclinical studies to clinical trials. From 1999 through 2002, there were only four trials for HD, but from 2011 to 2014, 28. More than 1,500 people who have HD are now participating in clinical trials, as well as many of their still-healthy relatives who are "pre-manifest," having inherited a mutation but not yet showing measurable signs or symptoms.
HD basics
Exactly two years ago, DNA Science introduced a family that illustrates an extreme of the multi-generational manifestation of HD, Juvenile Huntington's Disease: The Cruel Mutation. Karli and her father Karl died within 6 weeks of each other, and in this more recent post, Karli's sister Jacey discussed the juvenile form of the disease, which she has too.
Lisa Genova vividly conveys the rarity of HD with her own metric: the number of people with HD about equals the number of Red Sox fans who fit into Fenway Park in a scene in the book – about 37,000. The number who are "at-risk" – who have a parent with HD – is about five times that.
Each child of a person with HD has a 50% chance of having inherited the condition. If that happens, then the chance of developing symptoms is 100%, if a person lives long enough. Such "complete penetrance" is very rare. Symptoms typically begin in one's thirties, but changes in cognition (like the sudden inability to follow a favorite recipe) and mood (irrational anger) may creep in a decade or more earlier.
HD is an "expanding repeat" disorder. The gene (HTT) that encodes a protein called huntingtin (Htt) includes at its start a repeat of three DNA bases that encodes the RNA triplet CAG, which specifies the amino acid glutamine. Thirty-five or fewer "CAG repeats" is normal, but anything greater than 40 spells HD, with 36-39 copies a gray zone. Too many CAGs result in huntingtin protein that clogs brain parts that control movement and aspects of cognition.
Genova illuminates the meaning of extra repeats with a gut-wrenching scene in which Katie takes a black Sharpie and scribbles strings of 47 CAGs all over her bedroom wall. Her sister Meghan, a professional dancer, has just gotten the results of the genetic test that she took following required genetic counseling. Katie, sobbing, scrawls "the number of CAG repeats dancing inside the mind of her only sister."
How HD is different
The fact that HD is so unlike other conditions may explain why millions of research dollars haven't had much impact – yet. Three key distinctions are:

- Researchers can't compare the genomes and environmental exposures of people with the mutation who get the disease to people with the mutation who don't get the disease. They all get it.
- HD isn't due to a missing enzyme that can be replaced (like inborn errors of metabolism), a misfolded protein that can be re-folded (like cystic fibrosis), or a protein factor that can be supplied (like hemophilia). HD even differs from other triplet repeat disorders, such as fragile X syndrome, in which mangled proteins don't function. Instead, Htt has a new, "toxic gain of function."
- Having a working copy of the HTT gene doesn't compensate for or hide its evil twin on the other chromosome 4. That's why people with two HD mutations are no worse off than people with one, and why deleting the HTT gene has no effect.
These peculiarities suggest which therapeutic strategies are most likely to work.
Target DNA, RNA, or protein?
Gene therapy for HD doesn't make much sense. How would adding a functional HTT gene help, when it's already there? Blocking access to the extra genetic material seems the most logical approach to me, like redacting a phrase in a legal document with a heavy black mark, a biochemical version of Katie's sharpie.
Perhaps the most "druggable" part of the pathology isn't the gene or the protein, but the go-between RNA. A molecule of RNA riddled with extra-long repeats can contort in ways that can impart a hidden, second language, a powerful genomic subtext.
Repeats are scattered throughout our genomes, but extra-long ones can cause the messenger RNAs that seem to peel off of a gene to form secondary structures where G binds C within the molecule. This self-glomming can generate "R loops" where the RNA displaces one side of the DNA double helix, and ladder-like "RNA hairpins." A hairpin motif can trigger an immune response called PKR (protein kinase RNA-activated), which is usually deployed against viruses. The errant attack unleashes inflammation in the brain, while killing neurons.
Clinical trial updates
The fact that HD has no roster of drugs to choose from, as do cancer, HIV, diabetes, heart disease, and hepatitis C, certainly isn't due to lack of effort. Here are a few recent highlights that reveal efforts to think beyond the responsible gene. (For greater detail see the excellent Cure HD blog, by the pseudononymous Gene Veritas, who is at-risk).
Age of onset: A single nucleotide polymorphism (SNP) in the promoter of the HTT gene tracks with delayed onset if it is part of the mutant gene, yet an earlier onset if it is in the wild type (non-mutant) version. Kristina Becanovi, PhD, from the Karolinska Institute reported the findings recently in Nature Neuroscience. Although the study followed extreme cases, the discovery opens up "a smorgasbord of ideas for new therapies," according to co-author Ola Hermanson, PhD.
Genes other than HTT affect age of onset too. Jong-Min Lee, PhD, of Massachusetts General Hospital and co-workers discovered a gene on chromosome 15 that alters age of onset by up to six years, depending upon which variant a person has inherited.

Huntingtin Protein: Imaging can't capture Htt buildup well, but probing the protein's presence in cerebrospinal fluid (CSF) may provide a window to disease progression. Ed Wild, PhD, at University College London found that the closer pre-manifest patients got to symptom onset, the higher the level of Htt in their CSF. And the worse the symptoms, the more Htt.
Gene silencing: At least 14 companies are exploring variations on the gene silencing theme. ISIS Pharmaceuticals is about to launch a phase 1 clinical trial of an antisense oligonucleotide-based drug introduced into the spinal cord. Will CRISPR-Cas 9 one day replace the expanded HTT variant with one bearing a healthy 35 or fewer repeats?
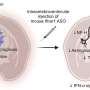
Stem Cells. An observational study called PRE-CELL is underway at the University of California at Davis' HDSA Center of Excellence (Vicki Wheelock, MD) and Institute for Regenerative Cures (Jan Nolta, PhD) to track biomarkers, symptoms, and imaging findings as HD progresses from diagnosis. After a year, patients can enroll in a phase 1 clinical trial that will deliver mesenchymal stem cells into the brain's striatum. The cells are altered to overproduce brain-derived neurotrophic factor (BDNF), which may save affected neurons.
Proteins that interact with HTT: Erich Wanker, PhD, from the Max Delbrück Center for Molecular Medicine and colleagues report in the May Genome Research a computational approach that deciphers Htt's interactions. The tool compares expression profiles of brain-specific genes in patients who have symptoms to those of individuals who are pre-manifest. Of 13 identified "interactors," 7 are already drug targets. Among the other six one protein, CRMP1, is expressed only in the brain. In animal models excess CRMP1 decreases Htt aggregation and cell death, and too little is associated with increased aggregation and cell toxicity. In cells, CRMP1 slows Htt aggregation.
New drugs: The only FDA-approved drug to specifically manage HD's motor symptoms is tetrabenazine, which has adverse effects of suicidality and depression. Auspex Pharmaceuticals is testing a drug called SD809 that tackles the chorea (uncontrollable movements) without tetrabenazine's adverse effects.
PKR inhibitors are compounds that derail the immune response to RNA hairpins.

Another drug candidate, SEN0014196, is an inhibitor of a sirtuin, which is a protein type associated with aging. The drug, a protein deacetylase, alters transcription of HTT. In animal models it extends survival and dampens movements, and speeds clearance of Htt aggregates while not affecting normal Htt protein. But I found the Internet trail to vanish after 2012. SEN0014196, aka EX-527, is commonly used in epigenetics research, so if someone can update use in HD, please do.
There have been pharmaceutical disappointments. Clinical trials for coenzyme Q10 and creatine were halted due to lack of efficacy. But sometimes a seeming failure isn't due to an ineffective drug, but to inadequate study design. That may be the case for pridopidine, a drug deemed ineffective in a clinical trial that considered only a subset of motor manifestations. Teva Pharmaceuticals is giving it another chance by testing total motor ability, cognition, mood, and quality of life. Teva is also evaluating a drug called laquinimod that quells brain inflammation.
Love is the drug
Lisa Genova has accomplished the seemingly impossible in Inside the O'Briens. She's made the story of a family shattered by HD into something approaching positive. At the risk of a spoiler, I'll just say that the book reaches a pivotal point where the trajectory and tone change in a magical way, a way that can really help families.
I thought I knew a lot about HD, from my writing but also from spending four months as a hospice volunteer with a young man so sick that his body became unable to move at all, out of energy. Ray's story inspired my novel about stem cells, music, and HD. But Lisa Genova, a neuroscientist, has taught me so much more.
"Inside the O'Briens" tells not about how to die from HD, but how to live with it in a way that guides younger generations, while enabling elders to finally understand what was wrong with their parents and grandparents, their aunts and uncles. Some readers may quibble with the ending. I did at first. But then I realized that it is a perfect metaphor for living with this genetically unusual and personally devastating inherited illness.
More information: "A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease" Nature Neuroscience 18, 807–816 (2015) DOI: 10.1038/nn.4014
"Systematic interaction network filtering identifies CRMP1 as a novel suppressor of huntingtin misfolding and neurotoxicity" Genome Res. Published in Advance April 23, 2015, DOI: 10.1101/gr.182444.114