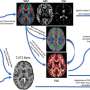
New research by Dr. Yan and her co-authors describes the cellular and molecular basis behind some autistic behaviors and suggests potential biomarkers and pharmaceutical targets. Credit: School of Medicine and Biomedical Sciences, University at Buffalo
Scientists at the University at Buffalo have identified the mechanisms behind a genetic mutation that produces certain autistic behaviors in mice, as well as therapeutic strategies to restore normal behaviors.
The research describes the cellular and molecular basis behind some autistic behaviors; it also suggests potential biomarkers and pharmaceutical targets.
Published May 28 in Cell Reports, the research was led by Zhen Yan, PhD, professor in the Department of Physiology and Biophysics in the UB School of Medicine and Biomedical Sciences.
The paper focuses on the loss of a gene called Shank3, an important risk factor for autism spectrum disorders (ASD). The researchers trace how this risk factor disrupts communication between neurons, leading to social deficits in mice. And, in their most important finding, they are able to reverse these neuronal disruptions, restoring normal behavior in mice.
Previous studies have shown that approximately 84 percent of people with a Shank3 deletion or loss-of-function mutation had an ASD. But just how this occurs has remained unknown.
The paper states that mice with a Shank3 deficiency exhibited "drastically reduced" interest in social stimuli, i.e., other mice, versus inanimate objects, suggesting "severe social deficits." They also spent significantly more time in repetitive self-grooming than normal mice.
The UB researchers found that the Shank3 deficiency plays a key role in how neurons communicate. It has a significant effect on the activation of the NMDA (n-methyl-D-aspartate) receptor, which is critical to learning and memory.
Yan explained that the Shank3 deficiency disrupts the trafficking of this receptor and its function at critical transmission sites in the brain. That disruption, they found, results from the dysregulation of actin filaments, which act as a kind of cellular "highway" in the brain's prefrontal cortex, the command center for "high-level" executive functions and a key region implicated in ASD.
"This research is the first to show that, in animals, abnormal actin regulation causes autism-like behaviors," said Yan.
"Actin filaments are very dynamic structures that are constantly being assembled and disassembled, processes controlled by numerous regulators," Yan explained.
When something upsets the equilibrium of actin filament assembly, key cellular functions fall apart.
"With Shank3 deficiency, we have found that the expression or activity of some actin regulators, such as cofilin, is altered," explained Yan. "This upsets the equilibrium of actin filament assembly, which, in turn, disrupts the normal delivery and maintenance of NMDA and other critical receptors."
The result is a very significant effect on the functional plasticity of the synapse, which, in turn, leads to the manifestation of some autistic behaviors.
In its most dramatic finding, the researchers found they were able to reverse this process, restoring normal behaviors in the Shank3-deficient mice, once the activity of cofilin or other regulators was returned to normal. This, in turn, restored actin dynamics at cortical synapses, allowing for the normal trafficking and functioning of NMDA receptors.
"Once actin filaments and NMDA receptors returned to normal, we observed a robust and long-lasting rescue of the social interaction deficits and repetitive behavior in the Shank3-deficient mice," said Yan. "Our results suggest a promising therapeutic strategy for treating autism."
The researchers are seeking funding to continue their work developing potential biomarkers and treatments for autism.
Journal information: Cell Reports
Provided by University at Buffalo