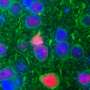
Blue is the nucleus, cyan is hUPF1, green is TDP43, red is the cell body in this image. Credit: Sami Barmada
Scientists from the Gladstone Institutes and the University of Michigan have identified a cellular mechanism that can be targeted to treat ALS. The researchers revealed that increasing levels of a certain key protein successfully protected against cell death in both genetic and sporadic versions of the disease. What's more, treating this pathway may also have implications for frontotemporal dementia because many of the same proteins are involved.
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a debilitating neurodegenerative disorder that leads to paralysis and death due to the loss of motor neurons in the brain and spinal cord. A primary feature of ALS is an accumulation of the protein TDP43, too much of which is toxic to cells. In the current study, published in the journal PNAS, the researchers identified another protein, hUPF1, that keeps TDP43 in check, thereby preventing cell death.
"TDP43 is a 'Goldilocks' protein: too much, or too little, can cause cellular damage," says first author Sami Barmada, MD, PhD, an assistant professor of neurology at the University of Michigan Medical School and former post-doctoral fellow at Gladstone. "Over 90% of ALS cases exhibit TDP43-based pathology, so developing a treatment that keeps protein levels just right is imperative."
Previous investigations had identified hUPF1 as a potential therapeutic target for ALS, but it was unclear how this protein prevented cell death. In the current study, the scientists tested hUPF1's ability to protect against neurodegeneration using a cellular model of ALS. They discovered that genetically increasing levels of hUPF1 extended neuron survival by 50-60%. Digging deeper, the researchers revealed that hUPF1 acts through a cellular surveillance system called nonsense mediated decay, or NMD, to keep TDP43 levels stable and enhance neuronal survival.
This protective mechanism (NMD) monitors messenger RNA (mRNA). If a piece of mRNA is found to be defective, it is destroyed so that it cannot go on to produce dysfunctional proteins that can harm the cell. It now appears that NMD also helps control the levels of proteins, like TDP43, that bind to RNA and regulate splicing. Since hUPF1 is a master regulator of NMD, altering it has a trickle-down effect on TDP43 and other related proteins.
"Cells have developed a really elegant way to maintain homeostasis and protect themselves from faulty proteins," says senior author Steven Finkbeiner, MD, PhD, a senior investigator at the Gladstone Institute of Neurological Disease. "This is the first time we've been able to link this natural monitoring system to neurodegenerative disease. Leveraging this system could be a strategic therapeutic target for diseases like ALS and frontotemporal dementia."
The scientists say the next step is to develop a drug that can target NMD—by manipulating hUPF1 or through other proteins that affect this system—to influence levels of TDP43 and protect neurons.
More information: Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1, PNAS, www.pnas.org/cgi/doi/10.1073/pnas.1509744112
Journal information: Proceedings of the National Academy of Sciences
Provided by Gladstone Institutes