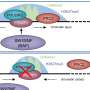
Human disease epigenomics 2.0
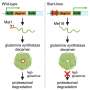
 includes transcription, cell subtype proportions and DNA sequence variability. As a further intriguing possibility, if DNA sequence variation causes unrecognised cell subtype lineage commitment effects (as lineage quantitative trait loci, linQTLs), it would follow that the presumed effects of DNA sequence variability upon gene expression and DNA methylation (eQTLs and mQTLs) may instead reflect effects upon cell subtype proportions. Credit: Greally Lab CC BY")
The study of how genes are regulated and how their regulation affects human disease has the potential to generate insights into mechanisms that aren't based on variation in DNA sequence, and could even show that temporally remote events can be "remembered" by the cell. Currently the method used by epigeneticists to examine these regulatory processes is an epigenome-wide-association study (EWAS). However, it is increasingly clear that the isolated EWAS is not sustainable as a robust means of gaining desired insights, and needs to be re-thought substantially. The human disease epigenomics 2.0 approach is a way of thinking about increasing the interpretability and value of these studies.
It's worth generalizing about the typical EWAS. These studies are usually designed as cross-sectional, comparing affected cases and unaffected controls at one time point rather than longitudinally over time. As another general rule, the same type of tissue or cells is usually compared in all individuals, recognizing that different cell types have different patterns of epigenetic and transcriptional regulation [1]. DNA methylation is the regulatory process almost universally studied in EWASs.
Epigenetic studies are mostly focused on testing the abundant 5-methylcytosine (5mC) modification, where a methyl group is attached to a carbon at position 5 in cytosine, but with a variable contribution of the minor 5-hydroxymethylcytosine (5hmC) modification where a hydroxyl group is added to the methyl group at the position 5. At present, most assays used do not discriminate 5hmC from 5mC [2], so 'DNA methylation' studies are generally measuring a combination of both 5mC and 5hmC. An increasingly wide range of human phenotypes is being tested for epigenetic dysregulation [3], based on the idea that a change in DNA methylation at the same site(s) in multiple affected cases when compared with controls is indicative of cellular changes characterizing the disease phenotype.
There is, however, growing concern that EWASs are not delivering reliable results, due to our recognition that DNA methylation is influenced by a number of factors. While any genome-wide assay is subject to technical and experimental variability, DNA methylation is also influenced by a number of biological influences. If the people studied have differences in the relative proportions of subtypes of cells in their samples from which DNA is extracted, that can affect the overall DNA methylation pattern generated [4, 5]. If a locus in the genome is transcribed to form RNA in some people and not others, this also has the potential to change DNA methylation at that locus [6, 7]. The normal differences that exist in DNA sequence between individuals represent an especially strong influence, accounting for between 22-80% of DNA methylation differences between individuals [8-10]. Analytically, there are some measures being taken to diminish the effects of cell subtype heterogeneity in particular [11], but less progress with the other sources of variability. If an EWAS has not tested for the contribution of major sources of variability, we cannot interpret the reason for any observed DNA methylation changes with any confidence.
There is not much we can do with current studies that were not designed to address these problems. We can, however, do better in our prospective design of new studies. A human disease epigenomics 2.0 era would involve the concurrent testing of the epigenome, transcriptome and genome, using cells in which the subtype composition can be determined, generating a rich dataset in which expression and methylation quantitative trait loci (eQTLs and mQTLs) allow insights into the effects of DNA sequence variability.
The potential then exists to use this characterization of interactions in control subjects as the foundation for understanding the deviations from these patterns in individuals affected by a disease, thereby defining epigenetic changes that are not accounted for by recognized confounding effects. Performing DNA methylation, transcriptional and genotyping studies in the same cells is certainly more expensive than testing DNA methylation alone. However, if isolated DNA methylation studies are not generating interpretable information, these would be cheaper but wasteful experiments. Furthermore, even the DNA methylation changes that are due to cell subtype, transcriptional or DNA sequence differences are potentially valuable as pathophysiological insights. A systematic change in representation of a cell subtype is potentially mechanistically contributory to a disease, as is a transcriptional difference between groups, while a DNA methylation difference attributable to an mQTL will have identified a genotypic association with the disease. These byproducts of the integrative human disease epigenomics 2.0 approach should be sought specifically, even if they do not test a starting hypothesis of independent epigenetic perturbations.
The first wave of EWASs has revealed DNA methylation changes associated with a wide range of phenotypes [3]. We now also appreciate that the ability to interpret these studies is constrained by our lack of information about known influences on DNA methylation. The isolated EWAS now needs to be supplanted by the more rigorous human disease epigenomics 2.0 approach, so that we generate fully interpretable data and robust insights into this exceptionally interesting alternative mechanism of human phenotypes.
More information: Won KJ, Zhang X, Wang T, Ding B, Raha D, Snyder M, et al. Comparative annotation of functional regions in the human genome using epigenomic data. Nucleic Acids Res. 2013;41(8):4423-32. Epub 2013/03/14. DOI: 10.1093/nar/gkt143. PubMed PMID: 23482391; PubMed Central PMCID: PMC3632130.
Plongthongkum N, Diep DH, Zhang K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet. 2014;15(10):647-61. DOI: 10.1038/nrg3772. PubMed PMID: 25159599.
Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;10(10):949-55. Epub 2013/10/01. DOI: 10.1038/nmeth.2632. PubMed PMID: 24076989.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. Epub 2012/05/10. DOI: 10.1186/1471-2105-13-86. PubMed PMID: 22568884; PubMed Central PMCID: PMC3532182.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2):R31. DOI: 10.1186/gb-2014-15-2-r31. PubMed PMID: 24495553; PubMed Central PMCID: PMC4053810.
Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39(1):61-9. Epub 2006/11/28. DOI: 10.1038/ng1929. PubMed PMID: 17128275.
Suzuki M, Oda M, Ramos MP, Pascual M, Lau K, Stasiek E, et al. Late-replicating heterochromatin is characterized by decreased cytosine methylation in the human genome. Genome Res. 2011;21(11):1833-40. Epub 2011/10/01. DOI: 10.1101/gr.116509.110. PubMed PMID: 21957152; PubMed Central PMCID: PMC3205568.
Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12(1):R10. Epub 2011/01/22. DOI: 10.1186/gb-2011-12-1-r10. PubMed PMID: 21251332; PubMed Central PMCID: PMC3091299.
Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7(8):e1002228. Epub 2011/08/20. DOI: 10.1371/journal.pgen.1002228. PubMed PMID: 21852959; PubMed Central PMCID: PMC3154961.
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6(5):e1000952. Epub 2010/05/21. DOI: 10.1371/journal.pgen.1000952. PubMed PMID: 20485568; PubMed Central PMCID: PMC2869317.
Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30(10):1431-9. DOI: 10.1093/bioinformatics/btu029. PubMed PMID: 24451622; PubMed Central PMCID: PMC4016702.
This story is republished courtesy of PLOS Blogs: blogs.plos.org.