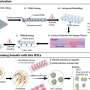
Gain-of-function p53 mutants are recruited by ETS family proteins to activate genes, including key epigenetic enzymes MLL1, MLL2 and MOZ. This, in turn, results in altered histone modifications genome-wide and may contribute to aggressive cancers associated with p53 mutations. Credit: Jiajun Zhu, Perelman School of Medicine, University of Pennsylvania
Aggressive cancer growth and alterations in gene activity without changes in DNA sequence (epigenetics) are associated with mutant p53 proteins, according to new research from the Perelman School of Medicine at the University of Pennsylvania. The international team describes their results and implications for difficult-to-treat cancers this week in Nature online ahead of print. The investigation was led by Shelley Berger, PhD, the Daniel S. Och University Professor in the departments of Cell & Developmental Biology, Genetics, and Biology, along with first author Jiajun Zhu, a PhD student in the Berger lab, and colleagues from the University of Toronto.
TP53 is the most frequently mutated gene among all human cancers. It encodes instructions for making a protein called tumor protein p53, simply p53, which normally suppresses tumors by regulating cycles of cell division. The p53 protein does this by keeping cells from growing and dividing too fast or in an uncontrolled way. When DNA becomes damaged, p53 elicits a series of protective responses to repair the cell or, if the damage is too severe, causes cell suicide. Mutations in the TP53 gene can undermine this normal function and allow cells with damaged DNA to continue to divide, leading to cancer.
In most cases, mutant p53 proteins are caused by a single mutation in one of the DNA building blocks, leading to a single amino acid substitution in the p53 protein. In addition to the loss of the normal p53 tumor-suppressing function, the substituted forms of p53 have also gained functions to promote cancer development in a more aggressive way.
To better understand how mutant p53 "gain-of-function" (GOF) works, the Penn team investigated cancer cell lines derived from patient tumors with different types of p53 GOF amino acid substitutions to see where these mutant forms of p53 actually bind in the cancer genome.
"We were surprised to find that mutant p53 binds to, and activates, a group of genes that comprises an epigenetic signature, especially those related to histone methylation and histone acetylation," Berger said. In particular, GOF p53 mutated proteins directly target genes encoding key epigenetic enzymes, including MLL1, MLL2, and MOZ.
In support of their observations, the team went to the The Cancer Genome Atlas (TCGA), a publicly available National Cancer Institute database of genetic characteristics of multiple types of patient tumors. Their analysis of TCGA data showed increased expression of the epigenetic regulatory MLL1, MLL2, and MOZ genes in GOF p53 tumors, compared to tumors with normal p53 protein or tumors without the p53 protein.
Gene expression is regulated by chemical modifications (including methylation and acetylation) on chromatin - histone proteins tightly associated with DNA. Certain chemical groups on histones allow DNA to open up, and others to tighten the chromatin. These groups alter how compact DNA is in certain regions of the genome, which in turn, affect which genes are available to be made into RNA (a process called transcription) and eventually proteins, the first step in many processes, including cell proliferation.
Normally, as an epigenetic enzyme, MLL1 puts a methyl group on the histone at a place that encourages transcription and favors cellular growth. They found that, for example, mutant p53 proteins tap into the MLL1 pathway, leading to genome-wide histone methylation changes, and therefore allowing for uncontrolled cell replication.
Altered epigenetic pathways have been implicated in various aspects of cancer, which might be a reasonable mechanism for explaining some uncontrolled cell replication, given the regulation of genome-wide transcription programs by epigenetic-related proteins. This finding provides the first evidence that GOF mutant p53 directly regulates key epigenetic factors.
To that end and most importantly, the team found that cancer cell proliferation was dramatically decreased by knocking down the gene for MLL1, which had the same result as decreased cell proliferation caused by knocking down the GOF p53 mutant gene.
"Now that we've determined we can inhibit cell proliferation by genetically inhibiting MLL1, specifically in p53mutant tumors, we also tested if we could inhibit MLL1 pharmacologically," Berger said. "We found that these cell lines were exquisitely susceptible." By using drugs that target MLL1 activity, the team found similar inhibitory effects on the growth rate of cells with mutant p53.
"Our study reveals a new epigenetic mechanism underlying the progression of tumors with gain-of-function p53 mutations," Berger said. "These findings indicate that these types of cancer cells thrive on these specific alterations. Gain-of-function p53 tumor cells are unable to replicate with abandon when these regulators are knocked down or pharmacologically inhibited."
In addition to MLL1, MLL2 and MOZ, this study has revealed that mutant p53 targets many other genes encoding epigenetic regulators. From this, the researchers aim to develop combinatorial epigenetic therapies for treating individual cancers driven by GOF p53 mutations. These types of cancers include, but are not limited to, those in the pancreas, breast, brain, esophagus, head & neck—all severe cancers typified by their inaccessibility to treat and late-stage diagnosis.
More information: Gain-of-function p53 mutants co-opt┬аchromatin pathways to drive cancer growth, DOI: 10.1038/nature15251
Journal information: Nature
Provided by University of Pennsylvania School of Medicine