and Brielle (8) Kennedy, love anything Disney. And they have type II spinal muscular atrophy")
These beautiful sisters, Brooke (6) and Brielle (8) Kennedy, love anything Disney. And they have type II spinal muscular atrophy
I began writing about genetics decades ago, and the best thing about getting older is witnessing the development of targeted treatments for single-gene diseases that I never thought would happen. But it is happening, for cystic fibrosis, Duchenne muscular dystrophy, hemophilia, various inborn errors of metabolism, blindness, and many other conditions. The steps may be incremental for some conditions, but researchers are deploying a staggeringly diverse armamentarium of techniques and technologies to fight genetic disease, from recombinant DNA-based drugs, to enzyme replacement and substrate reduction treatments, to gene therapy, antisense compounds, microRNAs, gene silencing, RNAi, exon skipping, and small molecule approaches.
Now spinal muscular atrophy enters the arena of the possible.
SMA isn't one of the more familiar single-gene conditions. Some people first read about it recently, in an August 13 Facebook post from Katherine Myers:
"To the little boy at the science museum, I don't know who you are, but thank you for being amazing. You let my son play and engage with you. You helped him pick up balls from the floor when you saw that he could not. You didn't ask what was wrong with him or why he couldn't walk, you just saw him. Kaden is a lot like you, he is very curious and wildly smart. He wants to know how everything works. Thank you for helping him turn the lever when you noticed he was too weak to do it himself. You will probably never see this but just by being you, you make this world better."
Brielle Kennedy was diagnosed with SMA at 16 months – before that she'd seemed fine, just a little slow in reaching certain physical milestones. Brielle's diagnosis came soon after sister Brooke was born, just days before dad Eric went to Afghanistan with the Michigan National Guard. A month after Eric left, his wife Sarah had to phone to tell him that Brooke, too, had inherited SMA. Their activism and fundraising began instantly, and the family inspired Representative Fred Upton to sponsor the 21st Century Cures initiative to support research on devastating childhood diseases. The Congressman and the two little girls have become close. The girls' motto is, "I can and I will."
SMA basics
In spinal muscular atrophy (SMA), motor neurons in the spinal cord degenerate. Skeletal (voluntary) muscle loses function, producing weakness and impairing mobility. Also called "floppy baby syndrome," SMA results from mutation in the "survival motor neuron" gene (SMN1). Incidence of SMA is 1 in 10,000 in the US, and 1 in 50 people are carriers – that's 6 million people who have one copy of the mutation.
Variants of SMA are based on clinical severity. In type I (Wernig–Hoffman syndrome), symptoms begin before 6 months, with death from respiratory failure by age 2. Type II, the most common form, is intermediate severity, with onset before 2 years. That's what Brielle and Brooke have. Children can sit but not stand or walk alone, and may live through their twenties. In type III (Kugelberg–Welander syndrome), symptoms begin after 18 months and range from overall weakness to requiring a wheelchair. Life expectancy may be normal but weakness worsens over time. In all types, children start out life alert and happy.
As with many genetic diseases, a few individuals defy categorization. When I was a hospice volunteer I visited a 7-year-old who had type I SMA. She couldn't move or respond and was on a feeding tube. I'd read to her but she was unable to respond in any way. Her brother, age 3, also had SMA, so perhaps other genes exerted a slight protective effect for the siblings.
Echo genes
SMA is a classic autosomal recessive illness: two carriers pass the disease to each child with a theoretical frequency of 1 in 4. But the genetic set-up is unusual, because the human genome includes two SMN genes. Nearly all patients have mutations, typically deletions, in both copies of the SMN1 gene, which is on the short arm of chromosome 5. The chromosomal neighborhood is a bit unstable because it is riddled with DNA sequence symmetry, including similar genes and repeats. This confounds DNA replication into making two copies instead of one, like repeating a word word in a sentence. That could be how two versions of the gene arose.

NHGRI
SMN2 is the second SMN gene, and people have 0, 1 or 2 copies on each chromosome 5. The most common situation is SMN1 and SMN2 splayed out in opposite orientation, encompassing half a million DNA bases. SMN2 is a little like an understudy for SMN1, because having extra SMN2 gene copies blunts the severity of SMA. Cells make a little more SMN protein.
Each gene – SMN1 and SMN2 – consists of 9 exons, which are the regions that encode the amino acids of a protein. The exons are separated by introns, the noncoding regions that are spliced out when the DNA is transcribed into messenger RNA. The two genes differ in just one DNA base, in exon 7, but it's at a critical part of the gene called an exon splice enhancer, which controls how introns are removed. The tiny difference in SMN2 effectively silences its ability to make the protein, nearly completely but not quite.
But what if we could turn SMN2 back on in SMA patients? It's not often that nature provides a back-up.
That's the reasoning behind a drug candidate called ISIS-SMNRx, being developed by Isis Pharmaceuticals, a Carlsbad CA-based biotech company that has been around for many more years than the unfortunately-named terrorist group. I remember writing about Isis in the early 1990s. The company is partnering with Biogen, another big biotech that's been around a long time.
How SMNRx works
Dr. Richard Finkel, division chief of neurology and a specialist in neuromuscular disorders at Nemours Children's Hospital in Orlando, explained the strategy of SMNRx. "The SMN1 gene is present in all animals. Only humans carry the 'back-up' SMN2 gene, which provides the opportunity for targeted treatment when SMN1 is deleted or mutated."
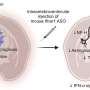
The targeted treatment for SMA is an antisense compound. That's a very short piece of a DNA-like molecule that forms complementary base pairs with (i.e. gloms onto) a selected part of a gene. For SMA, that's in the crucial exon 7 that alters intron splicing.
"The beauty of this strategy is that SMN2 does make a small amount of normal SMN protein, just not very much of it due to the single nucleotide change that alters splicing. The antisense oligonucleotide (the drug) targets an alternative splice site of SMN2 to promote inclusion of exon 7, that is usually excluded, and to shift the splicing emphasis to more full length transcript," Dr. Finkel explained.
The relationship of SMN1 and SMN2 on the same chromosome is a little like deriving the novels "To Kill a Mockingbird" and "Go Set a Watchman" from the single huge manuscript that author Harper Lee turned in to her editors back in the 1950s. They are each in a way cut from the same cloth, two stories culled from one, with the two genes more alike than the two narratives. "Go Set a Watchman" sat in silence for more than half a century, as did SMN2 until researchers figured out how to awaken it.
Great news so far
FDA has given SMNRx orphan drug status and fast track authorization. When the first clinical trial protocols were filed at the end of 2011, the best that could be hoped for was to show safety and perhaps slowing of progression. But it's much better than that. The company reported findings June 22 for the phase 2 clinical trial, and two phase 3 trials are ongoing, one for infants and one for older children.
In the phase 3 trials, 30 children who had received multiple doses of any of 4 escalating dosages in the phase 2 trial (to set baseline scores) are now receiving the highest dose every six months. The drug is given directly into the spinal cord. Progress is assessed every three months, and the company reported good news, so far, on May 15. The drug is not only safe, but seemingly effective at restoring some function.
Various "rating scales" are used to evaluate response. Some are used for several neuromuscular conditions, such as the "Six-Minute Walk Test" to assess number of steps taken on a treadmill in that time. Children who can't walk take the upper limb module (ULM) test, tweaked for SMA. Tailored to evaluating SMA is the Hammersmith Functional Motor Scale-Expanded (HFMSE) test. It assesses the best that a patient can do on a particular day, such as how long it takes to roll over and how long a child can hold a position, like sitting.
More than half of the children have improved in HFMSE scores, and performance on the other two measures increased too. "The natural course for children with untreated type II or type III SMA (is to) typically experience loss of muscle function that develops slowly and continually over time. A sustained increase of three or more points in HFMSE scores represents a significant departure from the natural course and is unusual for these children," said Dr. Darryl De Vivo, professor of neurology and pediatrics, Columbia University Medical Center.
Other treatments
Most genes don't have shadow copies to manipulate, but other approaches are targeting SMA too. At Nationwide Children's Hospital in Columbus, Ohio, Dr. Jerry R Mendell and colleagues are testing gene therapy for children with SMA1, using adeno-associated virus serotype 9, which is delivered intravenously.
Waiting in the wings are promising preclinical (animal) studies, which always continue even as treatments reach clinical trials. PTC Therapeutics has a small molecule drug that also shifts splicing of SMN2, boosting protein production. It's worked well in mice with a severe form of SMA, improving motor function, preserving motor neurons and the muscles they innervate, and extending life. (The mice are genetically modified and bred to have human SMN2.)
Kaden's mom's Facebook post has been viewed 10 million times and logged 230,000 likes. And the Kennedy family has told their story everywhere. Let's hope that the next time SMA is in the news, it's because of the treatment that looks so promising.
Provided by Public Library of Science
This story is republished courtesy of PLOS Blogs: blogs.plos.org.