Study pinpoints driver, potential target in aggressive pediatric leukemia subtype

A University of Colorado Cancer Center study scheduled for Feb. 18, 2016 online publication in the journal Cell Reports models Early T-Cell Precursor Acute Lymphoblastic Leukemia (ETP-ALL), discovering inactivation of the gene EZH2 as a driver and inroad to a potential therapeutic target in the disease.
"About 15 percent of acute lymphoblastic leukemia in children arises from T-cell precursors. We know that patients with T-ALL need more therapy upfront, we have to hit it harder. When this form of the disease relapses, or does not respond to established therapy, it's very hard to treat. The ETP-ALL subtype of T-ALL quite frequently appears to behave in this problematic fashion," says Tobias Neff, MD, investigator at the CU Cancer Center and pediatric oncologist at Children's Hospital Colorado.
A 2012 paper in the journal Nature, describing a large scale sequencing study of pediatric ETP-ALL, showed a complicated mix of genetic alterations with two major results: ETP-ALL shows frequent mutations in molecules mediating growth and survival signals, and more than 40 percent of ETP-ALLs have inactivating alterations of the gene EZH2. Loss of EZH2 function can in turn inactivate what is called Polycomb Repressive Complex 2 (PRC2).
However, how the Polycomb Repressive Complex 2 loss of function mutations would help leukemia growth was unclear from the 2012 Nature study. The current study from the University of Colorado Cancer Center now sheds light on this issue.
ETP-ALL was previously known to have stem cell-like gene expression. Neff, along with co-first author Etienne Danis, PhD, research associate in the Department of Pediatrics at the CU School of Medicine, and colleagues, now show that the differentiation from stem cells into more specialized blood cells partially depends on EZH2/PRC2, directly linking PRC2 loss of function to a stem cell-like gene expression profile.
"How exactly the stem-cell like gene expression profile contributes to the aggressiveness of ETP-ALL of acute leukemia is unknown, but we've known that these stem-like cells are associated with poor prognosis in acute leukemia," Neff says.
Another consequence of EZH2 inactivation is increased growth and survival signaling. In healthy tissues, Interleukin 6 signals cell growth, for example for the purpose of tissue repair after injury. In ETP-ALL, cells become especially sensitive to Interleukin 6, allowing these cells to grow far beyond healthy counterparts. The current study supports the concept that loss of Ezh2 enzyme spurs growth by increasing the expression of cellular receptors for Interleukin 6.
"We have two major features of the disease - stem-like cells and increased growth—and now we show an actor implicated in both, namely EZH2/PRC2," Neff says.
In fact, unpacking the mechanism of the gene EZH2 and the enzyme Ezh2 it encodes may have implications beyond ETP-ALL. The enzyme is an early player in a complex web of cellular communication that is also involved in a number of other diseases affecting children and adults, including neurofibromatosis, juvenile myelomonocytic leukemia and myelodysplastic syndrome.
Neff suggests that considering Ezh2 as an actor in many conditions illustrates a paradigm shift in the way modern science evaluates diseases, from a model in which pathologists define disease by its morphological appearance to a model in which disease is defined by its genetic signature. The diseases in this paragraph may look different in a microscope, but this work shows they may be closely related by their genetics.
"In addition to our specific finding in this disease, we are excited to now have a model that allows us to explore consequences of Ezh2 inactivation that may enrich our understanding of a number of other conditions with a similar set of genetic changes," Neff says.
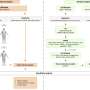
The group plans to use the newly developed mouse model to test therapies to combat the results of Ezh2 inactivation. Specifically, the paper shows that the existing drug ruxolitinib intercedes at an important link in the signaling chain that may be initiated by Ezh2 inactivation. With this link (namely JAK-STAT signaling) silenced by ruxolitinib, leukemia cells in the group's mouse model showed growth delay.
"Ruxolitinib is unlikely to treat the disease by itself, but this model will help us test possible drug combinations that could eventually benefit ETP-ALL patients," Neff says.
In future studies, the group plans to use the advanced genetic and chemical screening capabilities within the University of Colorado system to test the activity of many drugs against cells with inactivated Ezh2.