February 24, 2016 feature
Express this: Gene-specific transcription in humans linked to long-range connectivity of surface brain layers
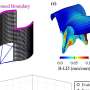
 Correlations between pairs of 114 brain regions in terms of their transcriptional profiles for the HSE genes. Correlations are averaged across individuals (n = 6). Complete black rows are brain regions that were not sampled by any individual. (B) Correlation matrix showing coupling patterns across the same 114 regions, measured by fcMRI at rest [adapted from Buckner et al. (27)]. Regions in both matrices are arranged such that those that belong to the same fcMRI network are grouped together. (C, Left) Surface representation of the 17-network parcellation used to group brain regions in A and B [adapted from Yeo et al. (31)]. White asterisks are locations of regions shown in the polar plot shown on the right. (Right) Polar plot showing transcriptional profile correlations between the auditory cortex region and surrounding regions, as well as distant occipital regions. Note the higher similarity of transcriptional profiles between somato/motor regions and occipital visual regions than to neighboring association cortical regions. Credit: Krienen FM, et al. (2016) Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain. Proc Natl Acad Sci USA 113(4): E469-E478.")
(Medical Xpress)—One of the most fascinating and fast-growing areas in neuroscience is evolutionary neurobiology - an interdisciplinary scientific research field at the intersection of neuroscience, evolutionary biology, and – after the so-called neo-Darwinian synthesis in the 20th century – comparative neuroanatomy and developmental genetics. Recently, a collaboration of scientists at The George Washington University, Washington, DC, Massachusetts General Hospital, and The National University of Singapore drew on an even wider range of disciplines, adding graph theory, molecular neurobiology, human cognitive neuroscience, and phylogenetic comparative brain evolution to determine if the expansion of the brain's cortical surface and associated long-range cortical connections in the supragranular layers of the cortex might be related to changes in underlying molecular architecture and thereby related genetic expression. They demonstrated that transcriptional expression of certain genes reflects the classic subdivision of the cerebral cortex into different types (sensory/motor, paralimbic, and heteromodal association), and that while the genes that best distinguish these subdivisions are selectively enriched in upper layers of the cortex in humans but not in mice, the researchers noted that the question of whether their findings apply only to humans or are common to other great apes or primates remains a hypothesis to be tested in future work.
Dr. Fenna Krienen (now in the McCarroll Lab at Harvard Medical School) and Medical Xpress discussed the paper that she and her colleagues published in Proceedings of the National Academy of Sciences. "Determining whether the expansion of the cortical surface (and the associated emergence of long-range connectivity networks) might be accompanied by molecular architecture changes is a tricky problem on a number of levels," Krienen tells Medical Xpress. "First, because in essence we're trying to characterize the end result of millions of years of evolution, which in the case of our species seems to have selected for extremely large brains in general and a disproportionately enlarged cerebral cortex in particular. If you look at the skulls of the early mammals, they were all quite small – perhaps the size of a mouse brain." She also notes that in today's mammals, a select number of cortical areas – that is, regions of the cerebral cortex specialized for sensory or motor function – are conserved in most or all living species. In humans, however, those conserved areas make up a small fraction of the cerebral cortex. In contrast, Krienen points out that the cortical areas that are most expanded in humans do not appear to be conserved and in fact also seem to support higher cognitive functions, such as language, problem solving, and imagining counterfactual scenarios.
"Therefore, the immediate question is," Krienen continues, "what's different – structurally, molecularly, and in terms of connectivity – about those regions?" The scientists' first challenge, she explains, was that connectivity between expanded cortical regions may not be able to be modeled by, for example, a mouse or nonhuman primate system because some of these regions may not exist in those species. "You can't study long-range connectivity directly in human brains, but you can estimate it from noninvasive techniques like MRI neuroimaging. When we did that, we saw a curious thing you don't really see in a small-brained species such as a mouse: the expanded parts of the cerebral cortex are located far apart from each other across the cortical surface, yet they seem to be strongly interconnected." Since in a mouse connections tend to be local, their questions then arose, what kinds of molecular features distinguish these expanded portions of the human brain and how might they be related to these unusual connectivity properties?
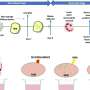
 Mean expression values for each of the 19 genes in the HSE gene set, plotted for seven networks. (Right) After mean expression is subtracted for each probe across cortical samples, relative differences in expression become evident across the seven networks. Credit: Krienen FM, et al. (2016) Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain. Proc Natl Acad Sci USA 113(4): E469-E478.")
There are other factors, Krienen notes, including molecular data from human brains being extremely scarce. "From a brain bank you may only be able to get a tissue sample from one or two brain regions at a time – but we needed to look at the molecular landscape across the entire cortex. That's why we, as a community, are very lucky that a few very valuable datasets have been made publicly available." Specifically, the scientists utilized a collection of six densely sampled postmortem human brains from the Allen Institute for Brain Science, Seattle, in which each brain was subdivided into hundreds of small regions, and the transcriptional profiles - identifying expressed genes – of each region was digitized. "From there," she says, "it was a matter of finding the structure in those transcriptional landscapes, and seeing how it compares with the brain networks we've been mapping using neuroimaging."
The researchers demonstrated that the transcriptional expression of certain genes reflects the classic subdivision of the cerebral cortex into different types – the initial challenge being the selection of genes to study. Since the team was interested in corticocortical connectivity – that is, connectivity between different areas in the cerebral cortex – and the cortex typically has a six-level columnar stack architecture (Layer 1 at the top, Layer 6 at the bottom), they were particularly interested in molecular changes to the upper layers. "Knowing in which layer a neuron resides is an important clue in figuring out what it connects with – and most neurons that connect to other cortical regions are found in Layers 2 and 3."
The paper also demonstrated Human Supragranular Enriched (HSE) genes – that is, those that are selectively enriched in upper cortical layers in humans relative to mice – as was shown in a 2012 paper by Hongkui Zeng and her colleagues1 which visualized at cellular resolution 1,000 HSE genes expressed in the cerebral cortex in mouse and human, showing that only a small number (19) of these genes had the unusual phenotype of being expressed in lower layers in the mouse but upper layers in human. "If this shift has something to do with the emergence of more corticocortical connectivity, then perhaps the transcriptional profiles of interconnected regions would be similar to each other for these particular genes."
Krienen notes that all of the in situ hybridization tissue sections used in the Zeng paper are available online at the Allen Institute's brain-map.org website. "However," she adds, "one challenge and outstanding question is whether this upper layer bias is a consistent feature of the human cerebral cortex, or whether it varies with brain area. The Zeng paper only probed two brain areas – the primary visual cortex and a region in the higher-order temporal cortex – so we still don't know whether these patterns hold for the hundreds of other areas in the human cortex. While our results – that distributed areas of association cortex have similar transcriptional profiles as measured by the microarray data – would predict that the pattern holds for different association areas beyond those found in the temporal lobe, this will have to be confirmed with future in situ hybridization or protein expression work."
The key insight that the researchers leveraged to address these challenges was bringing together two very large datasets that sampled very different information across the cerebral cortex – the set of postmortem brains for which gene expression for about 20,000 genes had been mapped, and a network map derived from fMRI of 1,000 living adults – which covered much or all, respectively, of the cerebral cortex. As a result, the scientists could look for broad commonalities and differences across the entire surface. "This was important," Krienen emphasizes, "in showing that distal association regions actually had similar genetic profiles for the HSE genes, as well as in allowing us to model other factors that affect transcriptional variation." One important factor, she illustrates, was the effect of spatial proximity: In general, proximate regions have more similar genetic profiles – so by having a densely sampled set of cortical regions, the team could show that the HSE genes predicted variation above the spatial proximity effect.
 Relative difference in distant–local degree connectivity measured by proportion of correlations that are within the local neighborhood versus distant correlations in resting state functional connectivity data [adapted from Sepulcre et al. (13)]. (B) Average distance score for 17 networks, listed along the group-averaged transcriptional similarity matrix for the HSE gene set. (C) Scatter plot showing the relationship between transcriptional similarity and degree connectivity score (from A) for all network pairs. Credit: Krienen FM, et al. (2016) Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain. Proc Natl Acad Sci USA 113(4): E469-E478.")
The paper states that molecular innovations of upper cortical layers may be an important component in the evolution of increased long-range corticocortical projections – an idea, Krienen says, that brings together a number of observations from this work and from other modalities. "We've known for a while that something different is going on in the upper layers of primate brains: Relative to rodents, they're disproportionately larger and more cell dense, and may also have unique cell types not found in other taxa. Again, we also know that the upper layers are the site of origin for most corticocortical connections, and that large primate brains have dense corticocortical connectivity networks."
The team put these observations together and thus raised the question of what had to change at a molecular level to allow this phenotype to occur. "Here we offer one possibility: that these HSE genes are among those associated with the emergence of more long-range connections. These may not be the only genes, but they provide a starting point for future work to understand how they might enable this form of connectivity."
The paper also presented results showing that the five categories of alternative gene sets tested did not consistently distinguish sensory and motor regions from paralimbic and association regions. "An important question was whether the results we found were truly unique to the genes we a priori selected," Krienen tells Medical Xpress. "To test this, we defined some alternative gene sets, several of which sought to vary only one of the HSE criteria at a time." For instance, they tested other genes previously shown to be uniquely expressed in the human brain but not specific to the supragranular layers. Another set comprised genes that are expressed in upper layers in both mice and humans.
"When none of these alternatives showed the strong association to connectivity networks, we went on to examine a large collection of curated gene sets. These sets, from the Broad Institute's Molecular Signatures Database (MSigDB), grouped together genes defined by their involvement in a biological process, coded for proteins in a particular cellular component, or were involved in a known molecular function. Our HSE set showed better association to the connectivity networks than all of these 1454 gene sets."
As to the planned next steps in her research, Krienen tells Medical Xpress, "The lingering question is exactly how the upper layers are remodeled in humans and other primates relative to rodents? While we know that the HSE genes are implicated in a number of different cell types, there's no smoking gun – namely, a cell type that explains the existence of long-range connectivity networks in humans. My next step is therefore to understand which cell types express these genes and how they may contribute to the remodeling of the upper layers."
More specifically, the issue is whether supragranular enrichment of the genes analyzed is unique to H. sapiens or common to other great apes or primates. "This will require us to move beyond human-mouse comparisons," Krienen agrees. "We diverged from rodents 80-90 million years ago, so comparative studies with other primates are necessary to determine whether the molecular innovation in the upper layers is a global trend of the primate lineage, or whether great apes or just humans made these modifications."
Other innovations Krienen is considering include drilling down to single cells. "Microarray and in situ hybridization are useful tools, but are limited when it comes to knowing exactly which genes are expressed in which cell populations – so right now I'm working with methods that allow me to profile gene expression in single cells. As with project we're discussing, I think this is the kind of thing that really benefits from a big data approach." To that end, the scientists are working with new high-throughput methods for profiling thousands of single cells using a single biological sample.
"In this study," Krienen concludes, "we drew on a number of research areas – namely, graph theory, genetics, molecular neurobiology, human cognitive neuroscience, brain evolution, and classical comparative neuroanatomy. Yet, we are still very limited in our ability to make sense of brain organization when we focus on only one or two species, so moving forward it will be exciting to see how the field of brain evolution – which still quite small – harnesses genetic tools and information in a broader range of species. That's really where I see the future going."
When asked by Medical Xpress, Krienen agreed that interesting ideas to consider might well be the 100m/s limit on axonal propagation speed as an adaptation that favored direct connection between distal brain areas over a serial relay network of shorter axonal/dendritic connections, thereby reducing redundancy and allowing parallelization of different types of information processing between neighboring regions and networks; increased brain energy efficiency derived from sparse but long connections versus all connections being short and serial; the possibility that evolution of long myelinated axons could enable a faster plasticity response by providing physically direct input of damage or dysfunction in a distal connected cortical area; and creation of a non-Linnaean taxonomy based on evolutionary neurobiology.
More information: Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain, Proceedings of the National Academy of Sciences (2016) 113:4 E469-E478, DOI:10.1073/pnas.1510903113
Related:
1Large-Scale Cellular-Resolution Gene Profiling in Human Neocortex Reveals Species-Specific Molecular Signatures, Cell (2012) 149:2 483–496, DOI:10.1016/j.cell.2012.02.052
© 2016 Medical Xpress