Stress pushes cells to die when gatekeeper of calcium use in mitochondria is dysfunctional
Malfunctioning mitochondria—the power plants in cells—are behind the damage caused by strokes, heart attacks, and neurodegenerative diseases, but little has been known about how to stop these reactors from melting down, destroying cells and tissue. Mitochondria also take up calcium, which regulates energy production. Now, online in Nature Communications, researchers at Thomas Jefferson University report important insights into how mitochondria are naturally protected against taking up too much calcium, which can force cells to die.
Their findings offer a novel avenue for creating drugs designed to stop mitochondria from demolishing cells during a time of stress, such as from a heart attack, says one of the study's senior authors, Professor György Hajnóczky, M.D., Ph.D., director of the MitoCare Center at Thomas Jefferson University.
"Mitochondrial failure causes much of the injury seen in the heart and brain during attacks and strokes, and inhibiting the brief overload of calcium that we see during these events could substantially reduce long term damage," he says. "This might become a novel and exciting treatment strategy."
Eventually, mitochondrial therapy could be developed to help treat neurodegenerative diseases, which are characterized by dysfunctional energy production, researchers say.
Research on mitochondria has recently undergone a renaissance, leading to establishment of the MitoCare Center at Jefferson University in 2014. Not only do mitochondria provide much of the energy for the life-sustaining cellular machinery but researchers have found they also help the cell make sense of signals from the environment that can change the cell's behavior. They are also realizing that mitochondrial dysfunction can signal the death of the cell, which underlies a wide range of disorders.
In this study, four research groups headed by Drs. György Csordás, György Hajnóczky, Jan Hoek and Erin L. Seifert, all from the MitoCare Center, used cell and animal studies to investigate the flow of calcium in and out of the squiggly looking cellular power plants.
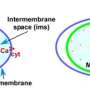
Several years ago, Dr. Hajnóczky and his team revealed the function of MICU1 (Mitochondrial Calcium Uptake 1). They found this protein controls the channel (MCU, mitochondrial Calcium Uniporter) that allows calcium to enter mitochondria. MICU1 acts as a gatekeeper preventing continuous intake but allowing calcium signals in to increase energy production.
The goal of the present research project was to understand the physiological and pathological role of MICU1. To find out what happens when mitochondrial calcium uptake is not properly regulated, the researchers developed the first animal model for loss of MICU1. They found that mice without MICU1 develop normally in utero, but die shortly after they are born. "These pups can't handle the stress involved in switching from intra-uterine life to extra-uterine life," Dr. Hajnóczky says.
They then developed adult mice in which MICU1 was removed only in the liver, an organ that can normally regenerate itself when damaged. The researchers discovered, in these mice, that the basic function of the liver was fine, but when the liver was exposed to stress (a portion was surgically removed), the liver could not regrow as it normally does because too much calcium flowed into cellular mitochondria, which triggered tissue death. "When stressed, and without the MICU1 calcium gatekeeper, the liver cells died," says Dr. Hajnóczky.
"The heart, the brain and the liver are very energy dependent, and can quickly get into trouble if mitochondria are dysfunctional," he says. "For example, a sudden loss of oxygen occurs in heart cells during a heart attack and in brain cells during a stroke also represent a stress for mitochondria. Due to the ensuing energy deficit, calcium accumulates in the cells and mitochondria are then flooded with calcium. If mitochondria cannot keep this calcium in check, they fall apart causing the cells to die.
"There are also indications of mitochondrial calcium dysregulation in a variety of neurodegenerative diseases, including Alzheimer's disease, Huntington's disease, and Lou Gehrig's disease," Dr. Hajnóczky adds. "These diseases don't necessarily begin in the mitochondria, but the damage they cause is amplified in these organelles."
"Inhibiting mitochondrial calcium uptake as a short term treatment could offer real promise for acute heart and brain attacks. It remains unpredictable in complex and chronic neurodegeneration whether a simple inhibition of calcium uptake would be effective. Rather strengthening both the sophisticated gatekeeping as well as the calcium signaling function of MICU1, might be sensible," he says.
The team has already begun to screen potential agents while continuing to tease apart the biological roots of MICU1 biology.
More information: Antony, A. N. et al. MICU1 regulation of mitochondrial Ca2 þ uptake dictates survival and tissue regeneration. Nat. Commun. 7:10955 DOI: 10.1038/ncomms10955 (2016).