In 2001, researchers developed a formula, or algorithm, that predicts whether a specific change in a gene sequence can result in harmful effects. While useful, the algorithm was slow; the computations underpinning these predictions used multiple central processing units (CPUs) and a significant amount of time. Now A*STAR researchers have adapted the algorithm to work on a graphical processing unit, a specialized electronic circuit that can process huge amounts of data in parallel.
The faster computational time has allowed the team to expand their "database of predictions" from just the human genome to include more than 200 additional organisms.
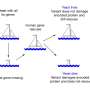
Similarities exist between the same genes of different organisms. Even so, individual organisms have differences in parts of their genomes when compared to other organisms of the same species. Some of these differences affect how proteins function and may lead to disease. By comparing genetic sequences, researchers are able to pinpoint disease-causing gene mutations. But this requires sifting through huge amounts of data.
The SIFT (Sorting Intolerant From Tolerant) algorithm predicts which changes in a gene — known as variants — could affect the function of the protein that gene encodes. Using SIFT, A*STAR researchers computed potential changes that can occur to gene sequences in humans to compile a database of predictions. Researchers provide SIFT with the gene variants they are investigating as a possible source of disease. SIFT then looks up the variants in its database of predictions. Variants that are predicted deleterious by SIFT are highlighted and may be considered worthy of further investigation.
Compiling SIFT's database for the human genome involved performing computations on multiple CPUs, which took about four minutes to analyse a single gene sequence.
"I had wanted to make SIFT databases for a lot more organisms, but making the human database took significant time," says systems biologist Pauline Ng from the Genome Institute of Singapore.
SIFT was adapted for use with a graphical processing unit to make faster predictions. This allowed the team to expand the scope of the algorithm's predictions to cover more than 200 other organisms. SIFT 4G, the updated algorithm, takes only 2.6 seconds to analyse a gene sequence compared to SIFT's four minutes.
The updated database and algorithm will not only facilitate the identification of disease-causing gene mutations but will help researchers understand the genetic variations that make some animal breeds or plants strains more robust or prone to disease.
More information: Robert Vaser et al. SIFT missense predictions for genomes, Nature Protocols (2015). DOI: 10.1038/nprot.2015.123
Journal information: Nature Protocols