Designer agent kills pain as well as morphine but may lack overdose risk

Investigators at the Stanford University School of Medicine and their collaborators at three other institutions have identified a novel compound that appears to exhibit painkilling power comparable to morphine but lacks that drug's most lethal property: respiratory suppression, which results in some 30,000 drug overdose deaths annually in the United States.
"This promising drug candidate was identified through an intensively cross-disciplinary, cross-continental combination of computer-based drug screening, medicinal chemistry, intuition and extensive preclinical testing," said Brian Kobilka, MD, professor of molecular and cellular physiology, and one of the senior investigators involved in the research.
Scientists at the University of California-San Francisco, the University of North Carolina and the Friedrich Alexander University in Erlangen, Germany, were also pivotal to the work, described in a study to be published Aug. 17 in Nature.
Kobilka credited Aashish Manglik, MD, PhD, a recent graduate of Stanford's Medical Scientist Training Program, as driving the study from the Stanford side. Manglik is one of the study's three co-lead authors.
The new compound's potential is enhanced by promising early signs, in mouse studies, that it may be less addictive than morphine and related drugs. While this reduced addiction potential remains to be demonstrated definitively in other animal studies, it's strongly suggested by, among other things, the experimental mice's indifferent attitude toward solutions containing the compound compared with otherwise identical solutions lacking it.
A drug with these characteristics would come as good news to physicians, patients and public-health authorities deeply concerned about a growing epidemic of addictive-painkiller abuse.
"Opium and its derivatives are perhaps the oldest drugs in the pharmaceutical formulary," said Manglik, who is now the School of Medicine's first-ever Stanford Distinguished Fellow, which enables him to have his own laboratory and independent funding. "There's some evidence that their use predates written history."
The hunt for a safer painkiller
A natural extract of the opium poppy, morphine was, in the 19th century, the first natural substance purified to homogeneity for medical use, Manglik said.
But respiratory suppression remains a general drawback of opioids, which in addition to morphine include the prescription painkillers codeine, oxycodone, oxycontin, hydrocodone and fentanyl as well as illicit drugs such as heroin. Designing a safer molecule required close collaboration between Stanford and scientists at three other institutions.
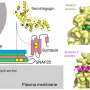
The new compound's identification made use of the three-dimensional structure of the mu opioid receptor determined by Manglik and colleagues in the Kobilka lab in 2012. The receptor, via which morphinelike drugs exert the bulk of their potent painkilling effect, is a member of a family of structurally similar cell-surface proteins found throughout the brain and spinal cord. When bound by morphine or one of its many natural or synthetic analogs, these receptors initiate signaling processes that alter the activities of other proteins inside the cells on which they sit.
Earlier work by other researchers established that morphine-resembling drugs' analgesic effect is brought about by a particular cascade of downstream chemical reactions (also known as a molecular pathway) set in motion when these drugs bind to the mu opioid receptor, while their respiration-suppressing effect is induced by another molecular pathway tripped off by the same binding event.
Safely reproducing morphine's benefits meant finding a way to separate those two effects. The trick was to activate the mu opioid receptor but not any of the other opioid receptors—and, having done so, to stimulate only the molecular pathway responsible for inducing analgesia and not the pathway responsible for respiratory suppression.
"The field had wondered whether a small molecule with just the right chemical features to trip off one pathway, but not the other, could be designed," said Manglik. Determining the mu opioid receptor's structure enabled detailed analysis of the receptor's binding pocket, into which opioids fit like a hand in a glove. This, in turn, propelled an interdisciplinary collaboration with scientists at UCSF, UNC and FAU.
Using a 'virtual medicine cabinet'
Manglik and Kobilka enlisted Henry Lin, PhD, then a graduate student in the lab of UCSF pharmaceutical chemistry professor Brian Shoichet, PhD. (Lin is a co-lead author and Shoichet is a co-senior author of the study.) After computationally screening about 3 million commercially available or easily synthesized compounds in a "virtual medicinal-compound cabinet" created by Shoichet's group, Manglik and Lin focused on 2,500 compounds that, computer simulations suggested, may bind to the mu opioid receptor. From those, they culled a few dozen that looked like especially good candidates for further inspection.
Lin and Shoichet focused on chemical structures that differed substantially from those of existing opioids, reasoning that they might bind to the receptor in ways that would stimulate beneficial but not detrimental downstream molecular pathways.
After testing 23 of these compounds and narrowing the field to seven, Lin and Manglik returned to the Shoichet group's online database, searched for similar compounds worth testing and found another dozen or so.
A dose of intuition
These compounds were sent to the laboratory of Bryan Roth, MD, PhD, a professor of pharmacology and of medicinal chemistry at UNC, who analyzed them further and found that one strongly activated the "good" downstream molecular pathway without significantly recruiting the "bad" pathway. Though promising, the compound was not sufficiently potent to work as a therapeutic. To optimize its properties, the group enlisted the help of Peter Gmeiner, PhD, chair and professor of medicinal chemistry at FAU. Gmeiner's group created numerous versions of the compound, and identified one that bound the mu opioid better than its predecessor.
An intuitive insight on Manglik's part led to a final tweak: the addition, in Gmeiner's lab, of a chemical feature called a hydroxyl group that would stabilize the molecule's "fit" inside the receptor's binding pocket. The resulting molecule, which the investigators named PZM21, had a mu opioid-binding strength about 1,000 times that of the compound in the original database from which it was derived.
Still more tests in the Roth lab showed that PZM21 not only didn't cause any significant activity in other opioid receptors but actually prevented activity in one of them, the kappa receptor, whose activation is associated with uneasiness and, sometimes, hallucinations. Both morphine and another opioid drug now in phase-3 clinical trials, oliceridine, trigger mild activity at the kappa receptor.
Experiments in mice by co-lead author Dipendra Aryal, PhD, a research associate in the Roth lab, bore out predictions of PZM21's analgesic efficacy—it was as powerful as morphine—and its benign character with respect to the suppression of breathing, compared with morphine. Given a choice between two chambers, one paired to an injection of a solution containing PZM21 and the other to an otherwise identical solution that lacked PZM21, the mice showed no preference for either chamber. By comparison, if one of the chambers is paired with morphine, mice are known to spend substantially more time in the morphine-paired chamber.
Other experiments performed in the Stanford laboratory of Gregory Scherrer, PhD, assistant professor of anesthesiology, perioperative and pain medicine and of neurosurgery, showed that PZM21 had no effect on mice bioengineered to lack the mu opioid receptor, confirming the compound's mechanism of action.
More information: Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G Levit A, Kling RC, Bernat V, Hubner H, Huang X-P, Sassano MF, Giguere PM, Lober S, Duan D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK. Structure-based discovery of opioid analgesics with reduced side effects. Nature, nature.com/articles/doi:10.1038/nature19112