Scientists develop therapeutic protein, protect nerve cells from Huntington's Disease
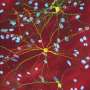
. The neuron in the center (yellow) contains an abnormal intracellular accumulation of huntingtin called an inclusion body (orange). Credit: Wikipedia/ Creative Commons Attribution 3.0 Unported license")
A new scientific study reveals one way to stop proteins from triggering an energy failure inside nerve cells during Huntington's disease. Huntington's disease is an inherited genetic disorder caused by mutations in the gene that encodes huntingtin protein. Approximately 30,000 Americans have mutant huntingtin protein which can impair energy-producing parts of nerve cells called mitochondria. The mutant protein destroys nerve cells and slowly chips away at a person's ability to walk, speak, and control their behavior. Xin Qi, PhD, assistant professor of physiology and biophysics at Case Western Reserve University School of Medicine has been looking for proteins that interact with mutant huntingtin to better understand the initial steps of Huntington's disease progression.
"Because mitochondrial dysfunction has been proposed to play an important role in the pathogenesis of Huntington's disease," said Qi, "we investigated the binding proteins of mutant huntingtin on mitochondria." His recent study published in Nature Communications characterized one protein, valosin-containing protein (VCP) that Qi's research team found in high abundance inside nerve cell mitochondria. Qi and colleagues discovered that VCP is recruited to nerve cell mitochondria by mutant huntingtin protein.
The researchers showed that mice with mutant huntingtin had mitochondria full of VCP, as did nerve cells donated by people with Huntington's disease. The VCP inside mitochondria only interacted with mutant, but not healthy huntingtin protein. According to Qi, "In Huntington's disease, the VCP-mutant huntingtin binding is greatly increased. This abnormal binding causes more VCP accumulation on the mitochondria," Nerve cells with VCP-mutant huntingtin interacting inside them became dysfunctional and self-destructed.
"We found that VCP is a key player in mitochondria-associated autophagy, a mitochondria self-eating process. Over-accumulation of VCP on mitochondria thus results in a great loss of mitochondria, which leads to neuronal cell death due to lack of energy supply." explained Qi. The researchers worked to identify ways to prevent VCP from heading to nerve cell mitochondria and interacting with mutant huntingtin protein once inside.
The team identified the regions of VCP and mutant huntingtin that were interacting. They cleverly designed a small protein, or peptide, with the same regions to disrupt the VCP-mutant huntingtin protein interaction. In nerve cells exposed to their peptide, VCP and mutant huntingtin bound the peptide instead of each other. Nerve cells exposed to the novel peptide had healthier mitochondria than unexposed cells. In fact, the peptide prevented VCP from relocating to mitochondria at all, and prevented nerve cell death.
Qi wanted to determine if the peptide had more than subcellular effects, and if it could be used therapeutically to prevent Huntington's disease symptoms. The researchers administered the peptide to mice with Huntington's-like disease and assessed mouse motor skills. Huntington's-like mice exhibit spontaneous movement including excessive clasping, poor coordination, and decreased lifespan. Mice treated with the novel peptide did not experience these symptoms and appeared healthy. Qi concluded that the peptide reduced nerve cell impairment caused by Huntington's disease in the animal model.
The study successfully countered harmful effects of mutant huntingtin and protected nerve cells in several models of Huntington's disease. According to Qi, the interfering peptide developed in the study "suggests a potential therapeutic option for treatment of Huntington's disease, a disease with no treatment available." The next step for the researchers will be to optimize the potentially therapeutic peptide for use in human studies.
More information: Xing Guo et al, VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington's disease, Nature Communications (2016). DOI: 10.1038/ncomms12646