A team of researchers at the Stanford University School of Medicine has used a gene-editing tool known as CRISPR to repair the gene that causes sickle cell disease in human stem cells, which they say is a key step toward developing a gene therapy for the disorder.
The team went on to demonstrate that the mended cells could make a functioning hemoglobin molecule, which carries oxygen in normal red blood cells, and then successfully transplanted the stem cells into mice. The researchers say the study represents a proof of concept for the repair of blood-borne genetic diseases, such as sickle cell disease and thalassemia.
A paper describing the findings will be published online Nov. 7 in Nature. Postdoctoral scholars Daniel Dever, PhD, and Rasmus Bak, PhD, are the lead authors; Matthew Porteus, MD, PhD, associate professor of pediatrics, is the senior author.
A painful and deadly condition
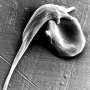
Sickle cell disease affects 70,000 to 100,000 Americans and millions globally, inflicting suffering and high health-care costs. Children born in high-income countries typically survive with the chronic disease, while those born in low-income countries typically die before the age of 5. The disease results from a single mutation in the gene that codes for one of the protein chains that make up the hemoglobin molecule. Hemoglobin is the main constituent of red blood cells and allows the cells to pick up oxygen from the lungs and drop it off in tissues throughout the body, from the brain to the muscles.
The sickle mutation causes the red blood cells to make an altered version of the hemoglobin that forces the red cell into a sickle shape when oxygen levels drop. The sickled cells tangle together, blocking blood vessels throughout the body and causing severe pain and disastrous health consequences.
The holy grail
Gene therapy, generally, has been the holy grail of gene-based therapeutics since the 1980s, with just a sprinkling of individual successes over the years. Increasingly, though, better techniques are raising hopes for practical therapies that can permanently cure genetic diseases like sickle cell.
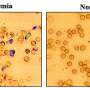
"What we've finally shown is that we can do it," said Porteus. "It's not just on the chalkboard. We can take stem cells from a patient and correct the mutation and show that those stem cells turn into red blood cells that no longer make sickled hemoglobin."
Porteus said that in previous work, he has targeted sickle cell genes with an older gene-editing technology but that the new CRISPR technology is faster and easier to work with. "We spent half a dozen years trying to target the beta globin gene using the old technology," he noted, adding that within one week of trying CRISPR, they had an editing tool that worked much better.
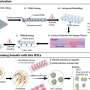
CRISPR is a combination of an enzyme that can cut a selected DNA sequence and a "guide RNA" that takes the enzyme exactly where you want to make the cut—in this case, at the sickle cell mutation. Once the mutated DNA sequence has been removed, other tools can help paste in a copy of the normal sequence.
The Porteus team started with human stem cells from the blood of patients with sickle cell disease, corrected the gene mutation using CRISPR and then concentrated the human stem cells so that 90 percent carried the corrected sickle cell gene. The stem cells are a particular type, called hematopoietic stem cells, that make blood cells. The team injected the concentrated, corrected hematopoietic stem cells into young mice.
"These stem cells have a property to be able to get from the blood system into the bone marrow where they then set up shop and start making other blood cells," said Porteus. When the team examined the bone marrow of the mice after 16 weeks, the corrected stem cells were thriving there.
The corrected red blood cells needn't replace all of a patient's original sickle cells, said Porteus. If the proportion of sickle cells is below 30 percent, patients have no symptoms of disease. And corrected cells have about a tenfold advantage over uncorrected cells, he said. That's because red blood cells afflicted with sickle cell tend to sickle and die after an average of only 10 days. In contrast, the corrected cells live the length of normal red blood cells, about 120 days. So the numbers of corrected cells rapidly outstrip those of uncorrected cells.
Is it safe?
Although gene therapy research has made great strides in recent years, it has yet to be widely deployed, and no CRISPR-edited genes have yet been tested for safety or efficacy in human clinical trials. Examples of potential problem include unforeseen immune reactions or altering the wrong sequence of DNA—so-called off-target effects. The effects of gene editing in general are impossible to predict.
As Porteus said, "The consensus in the field is that there's no one test we can do to prove that something is safe. We can't just say, 'Oh, just run this test, and that'll show if it's safe or not.' That test doesn't exist." Instead, he said, a series of different tests may each offer some insights about potential safety. For now, Porteus and his team found that their corrected human hematopoietic stem cells seemed to behave like normal, healthy human hematopoietic stem cells.
"We're excited about working to eventually bring this type of therapy to patients," said Porteus. "Stanford is building the infrastructure so that we can take our discovery in the lab and develop it so we can scale up the laboratory process to a process that will be needed to treat a patient. We hope to develop the entire process here at Stanford."
The team's work is an example of Stanford Medicine's focus on precision health, the goal of which is to anticipate and prevent disease in the healthy and precisely diagnose and treat disease in the ill.
More information: CRISPR/Cas9 Beta-globin Gene Targeting in Human Hematopoietic Stem Cells, Nature, DOI: 10.1038/nature20134
Journal information: Nature
Provided by Stanford University Medical Center