Genomic sequencing illuminates recent Shigella outbreaks in California

In a study that could have significant impact on how disease outbreaks are managed, researchers at UC Davis and the California Department of Public Health (CDPH) have sequenced and analyzed genomes from Shigella sonnei (S. sonnei) bacteria associated with major shigellosis outbreaks in California in 2014 and 2015.
The results offer new insights into how the bacteria acquired virulence and antibiotic resistance genes, as well as the California strains' relationships to other strains around the world. This was the first major, whole-genome study of S. sonnei strains found in North America. The research was published in the journal mSphere.
"If you have an outbreak and you want to know what is causing a particular problem, like antibiotic resistance, sequencing the genome can identify the genes involved," said Jonathan Eisen, professor with appointments in the Department of Medical Microbiology and Immunology and the Department of Evolution and Ecology at UC Davis and a collaborator on the study.
"Eventually, we should be able to sequence whole genomes of bacteria to support patient care," he said.
One of four Shigella species, S. sonnei is responsible for most shigellosis outbreaks and can cause abdominal pain, diarrhea and other gastrointestinal problems. Each year, shigellosis causes around 500,000 infections, 6,000 hospitalizations and 70 deaths in the U.S.
The investigators from the Microbial Diseases Laboratory at CDPH sequenced the genomes of 68 isolates, including samples from the recent California outbreaks and historical strains from California, Asia and elsewhere. They also tested for antibiotic resistance.
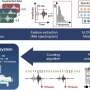
The team found two clusters in these outbreaks: one that primarily struck San Diego and the San Joaquin Valley and one more localized to the San Francisco Bay Area.
The San Diego/San Joaquin strain has been in California since at least 2008. However, some of the isolates had been infected with a bacteriophage (a virus that attacks bacteria) that carried a Shiga toxin (STX) gene found in the more virulent S. flexneri and S. dysenteriae.
"Shigella sonnei bacteria normally cause a less severe disease and are not known to produce Shiga toxin," said Dr. James Watt, Chief, Division of Communicable Disease Control at CDPH.
"The toxin gene was most likely acquired by Shigella sonnei via genetic exchanges with E. coli and other Shigella species. Discovering a functional toxin gene was concerning in this large outbreak. Finding this gene raises concerns that illness due to Shigella sonnei could become more severe in the future," Watt said.
By contrast, the strain that hit San Francisco lacked STX but contained genes that gave it resistance to the broad-spectrum fluoroquinolone class of antibiotics. The fluoroquinolone-resistance genes were similar to ones found in strains from Southeast Asia. These findings provide important clues to the strains' origins.
"We know these movements of DNA can be important for the spread of antibiotic resistance, virulence and pathogenicity factors," Eisen said. "Having the genome data from outbreaks allows us to try to figure out what happened."
The researchers believe similar studies might ultimately benefit patients. Understanding a pathogen's genetic variants could inform antibiotic choices and even help improve hospital procedures.
"If you can show that the transfer of antibiotic resistance genes came in response to some kind of treatment, you would certainly think about isolating people who were receiving that treatment, potentially sampling them more often or even changing treatments," explained Eisen.
Before that can happen, genomic sequencing must become a routine part of the nation's pathogen-surveillance model.
"It is clear, from a technical, economic, and scientific point of view, that we can and should be integrating more genome sequencing into infectious disease studies," Eisen said.
More information: Varvara K. Kozyreva et al. Recent Outbreaks of Shigellosis in California Caused by Two Distinct Populations of Shigella sonnei with either Increased Virulence or Fluoroquinolone Resistance, mSphere (2016). DOI: 10.1128/mSphere.00344-16