Researchers seek biomarker to assess spinal muscular atrophy treatment
Spinal muscular atrophy (SMA) is the leading genetic cause of death in infants, affecting 1 in 11,000 live births. As promising new therapies such as those directly targeting survivor motor neuron (SMN) are entering clinical trials for infants, children, and adults with SMA, researchers are searching for biomarkers in blood that can monitor their effectiveness. Investigators now report in the Journal of Neuromuscular Diseases that SMN levels in blood do not track SMN levels in motor neurons, and therefore are not an informative biomarker for SMN-modulating therapies that are delivered intrathecally. Beyond providing important information for SMA trial planning going forward, their results also highlight the importance of carefully validating specific biomarkers in a preclinical trial situation.
"This study highlights the importance of confirming the validity of potential biomarkers in a relevant, large animal model and will be useful in the design of future clinical trails," commented lead investigator Stephen J. Kolb, MD, PhD, Associate Professor, Department of Neurology and Biological Chemistry & Pharmacology, The Ohio State University Wexner Medical Center, Columbus, OH.
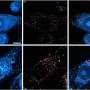
Investigators at The Ohio State University Wexner Medical Center explored blood SMN mRNA levels as a potential pharmacodynamic biomarker of an SMN-modulating therapy. The study took advantage of an important large animal model of SMA, the SMA piglet developed by the research group. The piglet models the anatomy and development of the nervous system of human infants well and provides a practical platform to perform these clinically relevant studies.
The SMA model pig was created using a gene therapy approach by knocking down the levels of pig SMN, followed by treatment with human SMN at early and late time points. This treatment was able to halt degeneration and it was hoped that monitoring blood levels of SMN would allow clinicians to follow the progress of this therapy and apply this marker to other potential therapies.
Researchers measured blood levels of pig SMN mRNA and human SMN mRNA in a porcine model of SMA longitudinally. However, they determined the blood levels of SMN are not altered when SMN levels are altered in motor neurons using a gene therapy approach delivered directly into the cerebrospinal fluid to target the central nervous system. "We found that there were no significant differences in the blood levels of pig SMN after knock-down and no evidence of human SMN after treatment. Interestingly, we also found that endogenous expression of pig SMN in the blood increases in the first month of life. There is potential significance to this observation if a similar increase occurs in human infants in this time period, because it is in the first three months of life that infants with type 1 SMA usually present clinically with progressive weakness," explained Dr. Kolb.
Over the last five years, promising results in preclinical models of SMA have led to a surge of clinical trials using small molecules, oligonucleotide-based therapies, and viral-mediated gene therapies. In December of 2016, Nusinersen (Ionis/Biogen) became the first drug to be approved by the U.S. FDA for the treatment of SMA. Another candidate, AVXS-101 (AveXis), is a gene therapy approach to increase SMN levels, which is in Phase I clinical trials.
"Although this biomarker was not useful in monitoring this particular therapeutic approach," noted lead author Chitra Iyer, PhD, "it is an exciting time to be in the field of SMA research since we can see our research translate from the bench to the bedside. In future, study of developmental changes in the fetal pig will provide valuable knowledge on SMN regulation in this critical period."
SMA is due to a defect in one gene (SMN1) and retention of another gene (SMN2), which result in low levels of the SMN protein that leads to progressive weakness and death.
More information: Chitra Iyer et al. SMN Blood Levels in a Porcine Model of Spinal Muscular Atrophy, Journal of Neuromuscular Diseases (2017). DOI: 10.3233/JND-170209