Credit: University of North Carolina at Chapel Hill School of Medicine
Among all the bad things that can happen to the brain when it is severely jolted – in a car accident, for example – one of the most common and worrisome is axon damage. Axons are the long stalks that grow out of the bodies of neurons and carry signals to other neurons. They are part of the brain's "wiring," and they sometimes grow to amazing lengths – from the brain all the way down to the spinal cord. But axons are thin and fragile. When the brain receives a strong blow, axons are often stressed past their structural limits. They either break or swiftly degenerate.
That much we know. But scientists haven't understood what happens next. What happens to the neuron when its axon goes south?
"Getting at the precise mechanisms of what happens after axon damage has been really challenging," says Anne Marion Taylor, PhD, an assistant professor in the UNC/NC State Joint Department of Biomedical Engineering. "But we think we've finally figured out a key part of what happens and why."
In a Nature Communications paper, Taylor and colleagues have revealed new molecular details of axotomy – when neurons are damaged or completely severed.
Shrinking dendritic spines, rising excitability
Scientists do know that a severed axon will cause a neuron to quickly lose some of its incoming connections from other neurons. These connections occur at short, root-like tendrils called dendrites, which sprout from the neuron's cell body, or soma.
Dendrites themselves grow tiny protrusions called spines to create actual connections, or synapses, with incoming axons. It's these dendritic spines that shrink in number following axotomy.

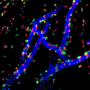
Taylor's lab uses a special chamber system so that when a neuron grows, its axon extends through a chamber, so that Taylor's team can conduct experiments. Credit: University of North Carolina at Chapel Hill School of Medicine
As it loses input connections, the wounded neuron also becomes more excitable: the neuron becomes more likely to fire signals down its truncated axon when stimulated to do so by other neurons. Neurons normally have a mix of inputs. Some are excitatory, pushing the neuron to fire; others are inhibitory, restraining the neuron from firing. Neurons with axons that have been truncated show a disruption of the normal excitatory/inhibitory balance in favor of excitability.
This enhanced excitability in the weeks and months following injury is thought to be largely an adaptive, beneficial response – a switch to a neuronal "seeking mode" like that seen in developing brains. This beneficial switch increases the chance that the neuron with the truncated axon can hook up with a new partner and continue to be a productive member of neural society.
"Neurologists know this," said Taylor, a member of the UNC Neuroscience Center. "It's why they promote physical therapy and retraining for people who suffer head injury. During this extended period of excitability, PT and retraining can help guide injured neurons along beneficial pathways."
But the injury-induced excitability in a neuron may cause problems too. A neuron can die from overexcitement (neuroscientists call this excitotoxicity). Neuronal hyperactivity after injury also may lead to intractable pain, muscle spasms, or agitation in the patient. In the days immediately after injury, doctors often treat brain-injury patients with drugs such as gabapentin designed specifically to suppress neuronal hyper-excitability.
What scientists haven't understood very well are the biological details, the hows and whys of dendrite spine loss and hyper-excitability. Those details have been elusive because of the spaghetti-like complexity of the brain, which makes it extremely difficult for a scientist to isolate a neuron and its axon for manipulation and analysis, either in a lab dish or a lab animal.
Several years ago, as a biomedical engineering graduate student at the University of California-Irvine, Taylor invented a device to help solve this problem. It's a microfluidic chamber with tiny grooves that trap individual axons from cultured neurons as they grow longer.
"The axons aren't able to turn around, so they just keep growing straight until they reach a separate compartment," Taylor says. "We can cut an axon in its compartment and then look at responses in the associated soma or dendrites without affecting axons in other compartments."

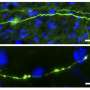
During a brain injury, neurons are damaged. After damage, they lose spines on their dendrites, as shown in the image on the right. Credit: Taylor Lab
A loss of inhibition
Taylor and her colleagues used the device in the new study to analyze what happens when an axon is severed. They found that events within the neuron itself drive the resulting dendrite spine loss and hyper-excitability. Signals originating at the site of injury move rapidly back along the remaining portion of the axon to the neuronal soma and nucleus, triggering a new pattern of gene activity. Taylor's team managed to block the neuron's gene activity to prevent the dendritic spine loss and hyper-excitability.
Taylor and colleagues analyzed how gene activity changed before and after axotomy. Multiple genes were altered following axotomy. The activity for one of these genes, encoding a protein called netrin-1, turned out to be sharply reduced. A separate analysis showed a similar drop in netrin-1 in affected neurons in rats whose axons from the brain to the spinal cord had been cut. Together, these results hinted that netrin-1's absence might be a major factor driving neuronal changes after axotomy.
When Taylor and colleagues added netrin-1 to axotomized neurons to restore the protein to normal levels – even two full days after severing the axon – they found that the treatment quickly reversed all of the dendritic spine loss and most of the hyper-excitability.
"The treated neurons more closely resembled uninjured controls," Taylor says. "This was a striking finding and we were surprised to find that netrin-1 normalized both the number of synapses and excitability, even when applied days after injury."
She added, "We're a long way off, but we really do hope to translate this netrin-1 finding into a new therapy. Ideally, it would do what gabapentin and related head-injury drugs aim to do, only better and more precisely."
Journal information: Nature Communications