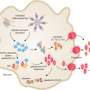
In hyperactivated macrophages, gasdermin D forms a limited number of pores -- enough to release IL-1 but not enough to kill the cell through pyroptosis. The macrophages stay alive and retain their phagocytic capacity, continuing to ingest microbes while stimulating memory T-cell formation via IL-1. Other proteins, like the cytokine TNF-alpha, are released through the classic secretion-signal pathway. Credit: Charles Evavold, Boston Children's Hospital
Researchers at Boston Children's Hospital have identified, for the first time, the molecule that enables immune cells to release interleukin-1 (IL-1), a key part of our innate immune response to infections. Findings were published online today by the journal Immunity.
IL-1, first described in 1984, is the original, highly potent member of a large family of cellular signaling molecules called cytokines, which regulate immune responses and inflammation. Several widely used anti-inflammatory drugs, such as anakinra, block IL-1 to treat such conditions as rheumatoid arthritis, systemic inflammatory diseases, gout and atherosclerosis. IL-1 is also a target of interest in Alzheimer's disease.
Yet until now, no one knew how it gets released by our immune cells.
"Most proteins have a secretion signal that causes them to leave the cell," says Jonathan Kagan, PhD, an immunology researcher in Boston Children's Hospital's Division of Gastroenterology and senior author of the new study. "IL-1 doesn't have that signal. Many people have championed the idea that IL-1 is passively released from dead cells: you just die and dump everything outside."
Recent work elsewhere at Boston Children's, for example, demonstrated that a protein called gasdermin D punches holes in the membranes of infected cells by forming pores. This releases IL-1 and other cytokines—but it also kills the cell, through an inflammatory cell-death program called pyroptosis.
In the new study, Kagan and first author Charles Evavold showed that gasdermin D also enables the release of IL-1 from living immune cells.
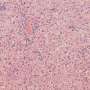
Dead macrophages (stained blue) don't make good infection fighters, unlike their living kin (green). It turns out macrophages don't need to die in order to release IL-1. Credit: Charles Evavold, Boston Children's Hospital
Stayin' alive
The idea of all immune cells sacrificing themselves to deliver IL-1 never seemed right to Kagan. Immune cells need to stay alive to carry out their other jobs, like stimulating the formation of memory T cells that provide long-lasting protective immunity.
Last year, Kagan, Ivan Zanoni, PhD, and colleagues reported in Science that IL-1 can be released from living dendritic cells, immune cells that present antigens to T cells. They showed that dendritic cells can enter a hyperactivated state in which they secrete IL-1, boosting their ability to rally memory T cells. But the question of how IL-1 gets released remained unanswered.
The new study involved hyperactivated macrophages, immune cells that engulf and ingest pathogens. "We found, in hyperactivated cells, that gasdermin forms enough pores to let IL-1 pass through, but not enough to kill the cell," says Kagan.
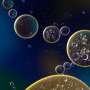
These living macrophages release IL-1, ingesting fungi (stained red) all the while. The bright green spots are inflammasomes, large protein complexes that detect danger signals and stimulate gasdermin D to form pores allowing IL-1 to pass through. Credit: Charles Evavold, Boston Children's Hospital
A path to better vaccine adjuvants?
The immune response is like a yin-yang: If it's too weak, you're susceptible to infections; if it's too strong, the system can tip toward autoimmune disease and, in the face of a serious infection, sepsis.
Gasdermin D inhibition is under study as a way of curbing inflammation-related diseases and sepsis. But Kagan's team is looking at the flip side, exploring gasdermin D as a target for stimulating IL-1 release from living, working cells to make vaccines more potent and long-lasting.
"For strong vaccination, you need to activate memory T cells, so you need a way to release IL-1 from living cells," says Kagan.
Interestingly, recent work shows that alum, the primary vaccine adjuvant in use today, works by activating inflammasomes. Inflammasomes are large protein complexes in cells that detect danger signals—and they're known to activate gasdermin D and pyroptosis.
Could gasdermin D be stimulated directly, in a way that does not kill the cell, to allow IL-1 out and dial up the immune response to vaccines? That's the idea Kagan's lab is exploring now. The lab is also exploring how a lipid called oxPAPC, which hyperactivates dendritic cells, supercharges memory T-cell activation by promoting IL-1 release through gasdermin D pores.
Journal information: Immunity
Provided by Children's Hospital Boston