Team develops technology to find optimum drug target for cancer

A KAIST research team led by Professor Kwang-Hyun Cho of the Department of Bio and Brain Engineering developed technology to find the optimum drug targets for specific types of cancer cells. The team used systems biology to analyze molecular network dynamics that reflect genetic mutations in cancer cells and to predict drug response. The technology could contribute greatly to future anti-cancer drug development.
There are many types of genetic variations found in cancer cells, including gene mutations and copy number variations. These variations differ in cancer cells, even within the same type of cancer, and thus, the drug response varies cell by cell. Cancer researchers worked to identify frequently occurring genetic variations in cancer patients, and in particular, the mutations that can be used as an index for specific drugs. Previous studies focused on identifying a single genetic mutation or creating an analysis of the structural characteristics of a gene network. However, this approach was limited in its inability to explain the biological properties of cancer induced by gene and protein interactions in cancer cells, which result in differences in drug response.
Gene mutations in cancer cells change the function of those genes and the ones that interact with it and its proteins. As a consequence, one mutation could lead to changes in the dynamical properties of the molecular network. Therefore, the responses to anti-cancer drugs by cancer cells differ. The current treatment approach that ignores molecular network dynamics and targets a few cancer-related genes is only effective on a fraction of patients, while many other patients exhibit resistance to the drug.
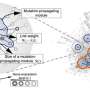
Professor Cho's team integrated a large-scale computer simulation using supercomputing and cellular experiments to analyze changes in molecular network dynamics in cancer cells.
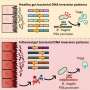
This led to development of technology to find the optimum drug target according to the type of cancer cells by predicting drug response. This technology was applied to the molecular network of known tumor suppressor p53. The team used large-scale cancer cell genomic data available from The Cancer Cell Line Encyclopedia (CCLE) to construct different molecular networks specific to the characteristics of genetic variations.

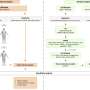
Perturbation analysis on drug response in each molecular network was used to quantify changes in cancer cells from drug response and similar networks were clustered. Then, computer simulations were used to analyze the synergetic effects in terms of efficacy and combination to predict the level of drug response. Based on the simulation results from various cancer cell lines including lung, breast, bone, skin, kidney, and ovary cancers were used in drug response experiments for compare analysis.
This technique can be applied in any molecular network to identify the optimum drug target for personalized medicine.
The research team suggests that the technology can analyze varying drug response due to the heterogeneity of cancer cells by considering the overall modulatory interactions rather than focusing only on a specific gene or protein. Further, the technology aids the prediction of causes of drug resistance and thus the identification of the optimum drug target to inhibit the resistance. This could be core source technology that can be used in drug repositioning, a process of applying existing drugs to new disease targets.
Professor Cho said, "Genetic variations in cancer cells are the cause of diverse drug response, but a complete analysis had not yet been made." He continued, "Systems biology allowed the simulation of drug responses by cancer cell molecular networks to identify fundamental principles of drug response and optimum drug targets using a new conceptual approach."
More information: Minsoo Choi et al, Network dynamics-based cancer panel stratification for systemic prediction of anticancer drug response, Nature Communications (2017). DOI: 10.1038/s41467-017-02160-5