Switching on survival signalling to drive drug resistance

Researchers at Queen Mary University of London have discovered that the loss of a single protein- PHLDA1- is sufficient for the development of drug resistance to a type of targeted therapy in endometrial and HER2-positive breast cancer cells.
Drugs that target specific pathways in cancer cells- so called targeted therapies- offer promising clinical benefits for cancer patients, with less severe side effects compared with more conventional chemotherapy agents. However, drug resistance- whereby cancer cells find ways to evade the effects of these drugs over time- limits the long-term clinical efficacy of these treatments.
A 'switch' for drug resistance
Receptor tyrosine kinases (RTKs) are key regulatory proteins involved in a variety of signalling pathways that control normal cellular processes, including growth, migration and survival. However, abnormal changes to these molecules are implicated in the development and progression of many types of cancer, making RTKs a popular target for cancer treatment.
Lead researcher Dr Richard Grose from Barts Cancer Institute at Queen Mary said: "Receptor tyrosine kinase targeted therapies often show excellent results in the clinic, but are undermined by the speed with which cancer cells develop resistance. We hope that, by understanding more about how resistance can develop, we can develop new approaches that improve the effectiveness of these drugs and offer better outcomes for cancer patients."
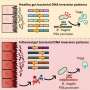
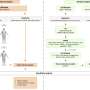
Using an RTK inhibitor to treat endometrial cancer cell models containing a mutated type of RTK, the team analysed the changes in gene expression and signalling activity as the cells shifted from drug-sensitive to a drug-resistant state. As drug resistance developed, downregulation of a protein called PHLDA1 was observed.
The team revealed that loss of this protein alone was enough to 'switch' on resistance in endometrial cancer cells. It appears that in drug-sensitive cells, PHLDA1 activity limits an important signalling pathway that cancer cells use to grow uncontrollably.
By rewiring themselves to reduce the level of PHLDA1, the cancer cells are able to find a way around the obstruction created by the drug. This results in a resurgence of pro-tumour signalling, allowing the cancer cells to thrive and become resistant to the effects of the RTK inhibitor.
To determine the translational potential of their findings, the team performed experiments to increase the level of PHLDA1 in the resistant cells. Rescuing PHLDA1 expression successfully re-sensitised the endometrial cancer cells to the RTK inhibitor.
This effect was not only observed in endometrial cancer cells. Breast cancer cells with high levels of HER2- an RTK that is overexpressed in 25-30% of breast cancers- also had significantly reduced levels of PHLDA1 as they became resistant to a HER2 inhibitor. Upregulation of PHLDA1 levels in these cells re-established a drug-sensitive state.
Looking at clinical data, where cancer patients had been treated with RTK inhibitors, they identified several studies that showed PHLDA1 expression was reduced following therapy, suggesting that the importance of these findings may extend beyond laboratory based models of cancer.
Future directions
The findings of this research, published in Cell Reports and majority funded by Cancer Research UK (CRUK), pave the way for future therapeutic approaches that may allow researchers to use this newfound knowledge to target drug-resistant cancer cells to improve treatment outcome for patients.
Having discovered this novel mechanism, the team are now investigating ways to safely re-express PHLDA1 in resistant tumours using drugs in the hope of re-sensitising cancer cells to RTK-targeted therapies. The team also aim to determine whether cells that downregulate PHLDA1 in order to become resistant to RTK inhibitors actually become vulnerable to other types of drugs.
More information: Abbie E. Fearon et al, PHLDA1 Mediates Drug Resistance in Receptor Tyrosine Kinase-Driven Cancer, Cell Reports (2018). DOI: 10.1016/j.celrep.2018.02.028