Researchers describe mechanism of protein accumulation in neurodegenerative diseases
Ludwig-Maximilians-Universitaet (LMU) in Munich researchers have characterized the mechanism that initiates the pathological aggregation of the protein FUS, which plays a central role in two distinct neurodegenerative diseases.
Aggregation of the DNA/RNA-binding protein FUsed in Sarcoma (FUS) in the cytoplasm of nerve cells is characteristic of two devastating and incurable neurodegenerative disorders. Amyotrophic lateral sclerosis (ALS) is a motor neuron disease that leads to progressive muscle weakness and ultimately to lethal paralysis, and frontotemporal dementia (FTD), the second most common form of dementia after Alzheimer's in those under 65. LMU biochemist Dorothee Dormann and her group are studying the molecular mechanisms that underlie both of these conditions. In a new study, they report that the aggregation of FUS observed in ALS and FTD is attributable to two distinct but functionally related molecular mechanisms. Their findings are reported in the leading journal Cell.
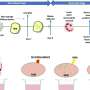
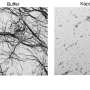
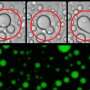
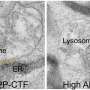
In healthy nerve cells, the FUS protein is transported into the nucleus, where it serves both as a DNA-binding transcription factor and as an RNA-binding regulator of transcript splicing. Strikingly, in the neurons whose function is affected in ALS and FTD patients, uptake of FUS into the nucleus is compromised. As a result, the protein accumulates in the cytoplasm of these neurons and coagulates into so-called stress granules. "Based on previous findings, we suspected that these granules might act as seeds to promote protein aggregation, ultimately leading to irreversible clumping of FUS," Dormann says. "However, the mechanism underlying the aggregation process was unclear." In the new study, she and her colleagues have now shown that FUS aggregation results from a liquid-liquid phase separation analogous to the gradual coalescence of oil droplets dispersed in an oil-water emulsion. The FUS protein forms liquid droplets, in which the protein can congeal into jelly-like clumps and finally solid aggregates.
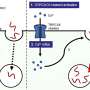
Dormann and her colleagues went on to show that, in ALS and FTD, these phase changes of FUS are promoted in two different ways. To get into the nucleus, FUS must interact with the nuclear import receptor Transportin. In the case of ALS, mutations in FUS significantly weaken the protein's ability to bind to Transportin. "Our results show that Transportin is not only responsible for delivering FUS into the nucleus, it also acts to stabilize proteins like FUS, which have a marked tendency to undergo phase separation," Dormann explains. This stabilizing function can be compromised in ALS patients by mutations in FUS. In patients with frontotemporal dementia, the FUS protein itself is normal. Instead, a small chemical modification on FUS that regulates its transport into the nucleus is defective. As the new study shows, failure to modify the protein not only affects its intracellular transport, but also promotes its phase separation and aggregation in the cytoplasm.
The results conclusively demonstrate that the aggregation of FUS is a central component of the process that leads to ALS and FTD. "Furthermore, accumulating evidence strongly supports the idea that other proteins implicated in the pathology of neurodegenerative disorders can form aggregates via phase separation," Dormann says. "We will now take a closer look at these proteins in order to understand how this process is normally suppressed, and how it might be modulated chemically."