Researchers develop 'cytological ruler' to build 3-D map of human genome
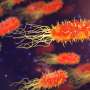
 produces a diffuse cloud of tyramide-containing molecules (red) that can label nearby DNA (blue). Sequencing the DNA can produce a map of the genome's organization. Credit: Chen et al., 2018")
It has been almost 20 years since the human genome was first sequenced, but researchers still know little about how the genome is folded up and organized within cells. In a paper to be published August 28 in the Journal of Cell Biology, researchers from the University of Illinois at Urbana-Champaign describe a new technique that can measure the position of every single gene in the nucleus to build a 3-D picture of the genome's organization.
The location of genes—whether they are close to the edge or the center of the nucleus, for example—may have significant effects on their activity, and a gene's position may therefore change as a cell develops or becomes diseased. Researchers can examine the position of individual genes using a microscope, but determining the position of every gene at the same time is impossible this way.
Yu Chen, Andrew Belmont, and colleagues from the University of Illinois at Urbana-Champaign have now developed a technique called tyramide signal amplification sequencing (TSA-Seq) that allows the distance of every gene from specific nuclear landmarks to be measured simultaneously. The study was carried out in collaboration with Jian Ma's group at Carnegie Mellon University and with researchers at the Netherlands Cancer Institute and Northwestern University Feinberg School of Medicine.
The TSA-Seq technique involves targeting an enzyme—horseradish peroxidase—to particular nuclear structures, such as the nuclear lamina that surrounds the nucleus or protein-containing granules called nuclear speckles that tend to be found in the center of the nucleus. The horseradish peroxidase then generates a highly reactive molecule called tyramide that can be used to label any DNA in the enzyme's vicinity. The closer a gene is to the enzyme, the more it will be labeled. So, when researchers subsequently sequence the cells' DNA, they can calculate how close each gene was to the nuclear structure tagged with horseradish peroxidase.
"TSA-Seq is the first genome-wide method capable of estimating actual distances of genes from particular nuclear subcompartments," says Chen.
Chen and colleagues tested their approach in leukemia cells and found that genes closer to nuclear speckles tended to be more active than genes closer to the nuclear lamina. Indeed, by examining the position of neighboring genes, the researchers were able to trace whole sections of chromosomes that looped out from the nuclear periphery toward speckles in the center of the nucleus. The function of nuclear speckles is unknown, but the regions of chromosomes close to speckles seem to be "hot zones" of gene activity.
"The logic of this nuclear organization remains to be determined, but our model would suggest that chromosome movements of just a few hundred nanometers could have substantial functional significance," Belmont says. A small shift in a gene's position so that it lies close to a nuclear speckle could be sufficient to dramatically enhance the gene's activity, for example.
The researchers say that the technique still needs to be improved, but they hope to use TSA-Seq to map the positions of genes in other cell types and examine how these positions change as cells develop or become diseased.
More information: Yu Chen et al, Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler, The Journal of Cell Biology (2018). DOI: 10.1083/jcb.201807108