Analysis of largest set of genomes from pregnant women reveals genetic links to disease, birth outcomes

Analysis of the world's largest set of genome data from pregnant women, totaling 141,431 expectant mothers from across China, has uncovered unsuspected associations between genes and birth outcomes, including the birth of twins and a woman's age at first pregnancy.
The analysis also allowed researchers to reconstruct the recent movement and intermarriage of different ethnic groups in China, and promises to help identify genes that make people susceptible to infectious diseases.
"It's amazing that this is even possible—that you can take these massive samples and do association mapping to see what the genetic variants are that explain human traits," said co-author Rasmus Nielsen, a professor of integrative biology at the University of California, Berkeley, who oversaw the computational analysis performed by researchers at BGI in Shenzhen, China.
It's even more amazing because the researchers sequenced, on average, only 10 percent of each mother's genome, relying on large numbers of poor-quality genomes so as to leverage cheaper tests to discover new genetic links.
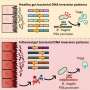
The mothers-to-be had provided blood samples to be tested for fetal chromosomal abnormalities, primarily Down syndrome. This technique, called cell-free fetal DNA testing, a form of non-invasive prenatal testing, is possible because mothers have DNA from their unborn child floating in their bloodstream. With rapid shotgun sequencing, labs can break up all the free-floating DNA in the blood and sequence just enough of the bits to diagnose Down syndrome.
Though not yet widespread in the United States, non-invasive prenatal testing is common in China: 70 percent of such tests worldwide have been performed in China. Sampling the mother's blood can be done early and risk-free, whereas standard prenatal testing in the U.S. involves amniocentesis or chorionic villus sampling, both of which require obtaining fetal cells from inside the uterus and risk harming the unborn child.
BGI was paid by maternity hospitals to conduct these tests, but obtained informed consent from each mother to also analyze the partially sequenced genomes for research purposes, maintaining anonymity. All the analyses were performed in China and the data is hosted in the China National GeneBank.
The data analysis revealed, for example, that variation in a gene called NRG1 is linked to a greater or lesser incidence of twins. One variant of the gene is more common in mothers with twins and is associated with hyperthyroidism, tightening a link between thyroid function and twinning that had previously been seen in mice.
A variant of another gene, EMB, was associated with older first-time mothers.
. Credit: BGI-Shenzhen")
The analysis also pulled out several genes that had not previously been associated with height and body mass index.
Perhaps most interesting, Nielsen said, is what sequencing of all the DNA in maternal blood tells us about viruses circulating through the body, and thus the link between viruses and genes that determine susceptibility to disease.
A variation in one gene, for example, was associated with a higher concentration of herpesvirus 6 in a mother's blood. Herpesvirus 6 is the most common cause of the relatively benign baby rash called roseola, but a high "viral load" correlates with more severe symptoms. People with Alzheimer's disease also have higher levels of herpesvirus 6 in their brains.
"Most people are infected by herpesvirus 6 at some point in their life, but some people seem to be less affected than others. We have now found a human genetic variant that helps control the severity of the infection," Nielsen said. "This is quite interesting because we don't know much about the genetic variants that control why some people seem more susceptible to viral infection and not others."
More correlations remain to be discovered. The BGI team to date has sequenced genomes from more than 3 million pregnant women, much of it accompanied by information on the mothers' and babies' health that can be used to find genetic associations.
"If you have these genotypes and compare them to phenotypes, that is, something you can measure, you can find genetic variants that explain human traits," said Xun Xu, a leader of the BGI team and the study's lead author.
Nielsen, Xu, Siyang Liu and other BGI colleagues will report initial findings from the analysis on Oct. 4 in the journal Cell.
Sequencing by imputation
To find genes associated with human traits—height and weight, for instance—researchers typically sequence thoroughly a small number of genomes—hundreds to thousands—and scan the genomes for variations in the sequence that are more common in people with the trait. The gold standard now is to sequence each genome 60 times to insure accuracy given inherent errors in the sequencing process. Even if each genome is sequenced a mere 20 times, which is good but not great, it still gets expensive.
The new study relies on only partial genomes—which are cheaper to get—but massive numbers of them. On average, about one-tenth of each mothers' genome was sequenced, because that is all that is necessary for a doctor to diagnose a chromosomal anomaly in the fetus. For example, Down syndrome, or trisomy 21, is caused by three rather than two copies of chromosome 21. A single cycle of sequencing is enough to determine whether some genes are 50 percent more common than normal, indicative of one extra chromosome.
But partial genomes can tell researchers a lot too, Nielsen said.
Think of reconstructing a lost book from thousands of error-prone copies, complicated by the fact that you have only about 10 percent of each copy. By looking for overlaps and inferring words from context—called imputation—you could reconstruct a lost manuscript.
In reconstructing partial genomes, scientists have another important data set: all the complete human genomes sequenced to date, with all their individual variations.
Proof that imputation using more than 141,000 partial genomes works is that the reconstructed geographical distribution in China of minority groups and the dominant Han Chinese reflect known population movements in the country over the last 100 years.
"Because the sample size is so large, we can get at recent population movements, including relocations as a consequence of China's governmental policies," Nielsen said. Many populations of Han Chinese in western China are more closely related to the populations of large cities on the East Coast, for example, reflecting relocation of large numbers of people into the sparsely populated countryside.
The researchers also found that many Chinese had genetic variants common among Indians, Southeast Asians and, along the route of the old Silk Road, Europeans.
Nielsen is currently working with his BGI colleagues to analyze the genomes of 1 million Chinese women who underwent non-invasive prenatal testing.
More information: Cell, DOI: 10.1016/j.cell.2018.08.016