Pharmacological therapy to fight a rare pediatric disease
Scientists from the University of Granada (UGR) have developed a new pharmacological therapy using a molecule that presents similarities with one of the precursors of coenzyme Q10 (CoQ10). The therapy is effective against a severe mitochondrial disease that targets mostly children, and for which there is currently no effective treatment.
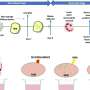
Published in EMBO Molecular Medicine, the study reports the effectiveness of this experimental treatment in an animal model with severe deficiency in CoQ, encephalomyopathy and premature death. The treatment involves the chronic administration of an analogue of one of the precursors that eukaryotic cells use to synthesize CoQ.
CoQ10is a molecule synthesized in cells and has key functions in cellular metabolism. Deficiency in CoQ10 is a syndrome with diverse clinical manifestations. Usually, patients that present neurological symptoms or those that manifest the multisystem variant do not respond to the conventional treatment with high doses of exogenous CoQ10 due to the low capacity of this molecule to cross biological obstacles and reach the nervous system.
The researchers assessed a therapeutic option that is effective for severe cases of deficiencies in CoQ10. The working hypothesis of the researchers was based on recent studies in yeasts and cell cultures generated by some international groups, including the UGR group.
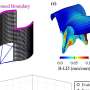
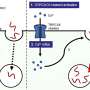
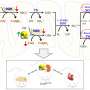
The results indicate that the therapeutic molecule is capable of modulating the multiprotein complex for CoQ biosynthesis, thereby stimulating endogenous CoQ synthesis. "But we have also seen, and this is even more striking, that the therapeutic molecule is capable of reducing the levels of intermediary metabolites of CoQ that can be toxic to the power plant of the cells," explains Luis Carlos López García, a researcher of the UGR Biomedical Research Center and one of the article authors.
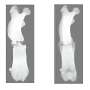
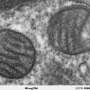
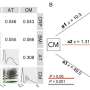
Consequently, these metabolic changes induce an increase in the capability of cells to produce energy, together with a reduction in the molecular and histopathological markers characteristic of mitochondrial encephalomyopathies.
The final result is an improvement of the phenotypic characteristics of the animal model with a very significant increase in life expectancy. While the untreated mutant mice reach a maximum of seven months of life, the treated mutant mice are able to reach up to 25 months of life, approaching the survival curve characteristic of healthy mice.
"The therapeutic results we have obtained are very important, so one of the medium-term objectives is to apply it in the clinic," Prof. López says. "There are some response mechanisms to the treatment that we still do not understand, so we are working on new experimental approaches that will help us to figure out these aspects, which should contribute to better understand the pathophysiology of mitochondrial diseases and to generate new therapeutic advances for this and other mitochondrial disorders. At the same time, we are testing other molecules with very interesting characteristics that may be important not only for the treatment of severe diseases but also as nutraceuticals for the healthy population or for people without severe pathological conditions."
More information: Agustín Hidalgo‐Gutiérrez et al. β‐RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9R239X mice, EMBO Molecular Medicine (2018). DOI: 10.15252/emmm.201809466