Scientists develop new mouse model of Hirschsprung's disease

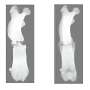
About one in every 5,000 babies is born without enteric neurons in the distal colon resulting in Hirschsprung's disease. Because of the missing neurons, contents of the gut cannot pass normally, resulting in constipation and enlargement of colon.
The condition is treated with a surgical removal of the affected gut part, but patients remain at high risk of enterocolitis, or inflammation of the gut. This is the major life-threatening complication of Hirschsprung's disease.
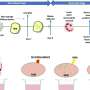
About half of the Hirschsprung's disease cases are caused by mutations in a gene called RET. RET is a receptor, a large protein molecule, located at the surface of the cell to receive signals from other cells. During development, a complex formed by two proteins called GDNF and GFRa1 binds to RET and activates signaling required for normal development of the enteric neurons.
For developing new treatments, animal models of the disease are most often a prerequisite. This work lead by Associate Professor Jaan-Olle Andressoo describes generation and characterization of the first viable mouse model of Hirschsprung's disease and associated enterocolitis with a defect in GDNF/GFRa1/RET signaling, thus representing most patients.
This is important, because so far, animal studies of Hirschsprung's disease have used model systems that represent a minority of the genetic mutations in Hirschsprung's disease. Using the new mouse model, scientists at the University of Helsinki shed further light on the chronology of events in enterocolitis. They found that mucin-producing goblet cells, a specific type of cell responsible for lubricating the inner surface of the gut, may be a potential target for preventative treatment.
Scientists also conclude that reduced expression of GFRa1 can contribute to susceptibility for Hirschsprung's disease. The new mouse model will serve as a useful tool for enhancing understanding of the disease and for defining treatment in the future.
More information: L Lauriina Porokuokka et al, Gfra1 under-expression causes Hirschsprung's disease and associated enterocolitis in mice, Cellular and Molecular Gastroenterology and Hepatology (2018). DOI: 10.1016/j.jcmgh.2018.12.007