")
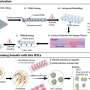
Figure 1.Mechanism of autophagy in dendritic cells. Credit: The Korea Advanced Institute of Science and Technology (KAIST)
Autophagy contributes to the homeostasis of a cell and recently another function of autophagy has been reported. A KAIST research team found that the autophagy of dendritic cells supports T-cell anticancer activity.
Autophagy is a process of maintaining cell homeostasis by removing cellular waste and damaged cellular organelles; nevertheless, its role in the presentation of phagocytized tumor-associated antigens remains vague.
Meanwhile, dendritic cells are the ones that recognize pathogens or cancer antigens, and induce immune responses in T cells. When cancer cells are killed by radiation or an anticancer drug, dendritic cells absorb and remove them and present antigens on their surface to transfer them to T-cells.
Professor Heung Kyu Lee from the Graduate School of Medical Science and Engineering and his team found that the autophagy of dendritic cells plays a key role in T-cell activation and they proposed the principles of enhancing anti-cancer effects.
Their experiments showed that T-cell activation of dendritic cells as well as anticancer immune response dropped when there is a deficiency of Atg5 (autophagy-related) in dendritic cells.
Interestingly, Atg5-deficient dendritic cells significantly elevated receptor CD36 on the surface of the cells, which increased the phagocytosis of apoptotic tumor cells yet restricted the activation of T-cells.
")
Figure 2. A role of autophagy in dendritic cells. Credit: The Korea Advanced Institute of Science and Technology (KAIST)
At this time, when introducing antibodies into the system in order to block the receptor CD36, the anti-tumor T-cell response increased substantially while tumor growth declined.
Professor Lee said, "This study allowed us to explore the role of autophagy in the anti-cancer immune response of T-cells. We look forward to developing targeted anti-cancer therapies using the receptor CD36."
This research was published in Autophagy.
More information: Dong Sun Oh et al. Autophagy protein ATG5 regulates CD36 expression and anti-tumor MHC class II antigen presentation in dendritic cells, Autophagy (2019). DOI: 10.1080/15548627.2019.1596493