How the enzyme lipoxygenase drives heart failure after heart attacks

Heart failure after a heart attack is a global epidemic leading to chronic heart failure pathology. About 6 million people in the United States and 23 million worldwide suffer from this end-stage disease that involves dysfunction of the heart, a change that clinicians call cardiac remodeling. Despite medical advances, 2 to 17 percent of patients die within one year after a heart attack due to failure to resolve inflammation. More than 50 percent die within five years.
Ganesh Halade, Ph.D., is seeking ways to delay or reverse this heart failure, which comes from non-resolved chronic inflammation. Over-activated leukocytes from the spleen that rushed into the heart muscle to remove dead tissue and start repairs are not adequately calmed and do not receive a "get out" signal.
So, learning the details of metabolic signaling that controls the immune responses—both during the acute inflammation after injury and the resolution thereafter—is important. Halade, a University of Alabama at Birmingham associate professor in the UAB Department of Medicine Division of Cardiovascular Disease, is working to discover which metabolic signatures are biomarkers for healthy physiology and which metabolic signatures are biomarkers for heart failure pathology.
This could permit the development of a prevention plan and precise, prognostic and personalized measures to delay heart failure.
This work follows his 2017 discovery that knocking out 12/15 lipoxygenase, or 12/15LOX, a lipid-modifying enzyme that competes with two other lipid-modifying enzymes, leads to increased survival in a mouse model of heart failure after a heart attack.

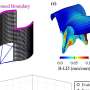
In a study now published online ahead of print in the journal Metabolism: Clinical and Experimental, Halade and colleagues detail the profound lipidomic and metabolic signatures and the modified leukocyte profiling that delay heart failure progression and provide improved survival in 12/15LOX-deficient mice. Only 6 percent of the 12/15LOX-deficient mice died in the progression of chronic heart failure, 56 days after heart attack, while 38 percent of mice with normal 12/15LOX had mortality due to heart failure or rupture.
Specifically, the researchers quantified changes in the metabolome, lipidome and immune profiles during acute heart failure, one day after heart attack, and during chronic heart failure, eight weeks after heart attack.
They found that the 12/15LOX-deficient mice biosynthesized the signaling molecules epoxyeicosatrienoic acids—also known as EETs or cypoxins—in left ventricle heart tissue after heart attack to facilitate cardiac healing. The lipoxygenase-deficient mice also had reduced amounts of the diabetes risk biomarker 2-aminoadipic acid and had profound alterations of plasma metabolic signaling of hexoses, amino acids, biogenic amines, acylcarnitines, glycerophospholipids and sphingolipids during acute heart failure. These changes are accompanied by delayed heart failure and improved survival.
"Future studies are warranted to define the molecular network of the lipidome and metabolome in acute and chronic heart failure patients," Halade said, though he notes this needs to be preceded by work with other animal models. "Collectively, our studies have discovered a novel link of LOX signaling between lipidomic and metabolic signatures in acute and chronic heart failure syndrome."
More information: Ganesh V. Halade et al. Lipoxygenase drives lipidomic and metabolic reprogramming in ischemic heart failure, Metabolism (2019). DOI: 10.1016/j.metabol.2019.04.011