UNC Lineberger’s Xian Chen, PhD. Credit: University of North Carolina at Chapel Hill School of Medicine
Precision cancer medicine requires personalized biomarkers to identify patients who will benefit from specific cancer therapies. In an effort to improve the accuracy of predictions about prognosis for patients with breast cancer and the efficacy of personalized therapy, University of North Carolina Lineberger Comprehensive Cancer Center researchers have developed a method to precisely identify individual patients who have aggressive breast cancer.
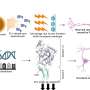
The new approach involves sorting and characterizing invasive breast cancer cells by epigenetic characteristics—a method that involves analyzing how particular regulatory proteins interact with DNA to control their expression—as well as by how the genes are amplified or abnormally expressed. The researchers reported in the Cell Press journal iScience they used this technique to identify potential new prognostic markers to predict distinct clinical outcomes for two major subtypes of breast cancer.
"This paper describes a ground-breaking multi-omics technology to discover drivers of proliferative and invasive breast tumors," said UNC Lineberger's Xian Chen, Ph.D., professor in the UNC School of Medicine Department of Biochemistry & Biophysics. "We think that eventually, these tools could help doctors better predict which particular patients have a good response, or acquire resistance to treatment."
Doctors often rely on information about tumor size, whether the cancer has spread and the tumor subtype to make treatment decisions. In addition to clinical subtypes of breast cancer, researchers have discovered molecular subtypes that have been used to help make treatment decisions. However, Chen argues that existing markers do not adequately distinguish breast cancer patient subpopulations with different clinical outcomes.
"Single 'omics' approaches, which rely on either genomics, transcriptomics, or proteomics alone, fail to dissect the heterogeneity that contributes to individual patients' variability in terms of their rates of tumor growth, metastasis, or susceptibility to anti-cancer therapies," he said. "Because biomarkers are not available to distinguish distinct patient sub-populations that are either responsive or resistant to particular drugs, doctors do not have all the tools they need to predict patient response to treatment and outcomes."
In their study, the researchers wanted to see if they could stratify patients beyond existing molecular subtypes. Their goal was to develop a method to determine which patients within a single subtype would develop resistance or invasive cancer. There are five major molecular subtypes of breast cancer, which are classified based on how genes are expressed in a tumor.
Chen and his colleagues analyzed luminal breast cancer and basal-like breast cancer, which is more commonly known as triple negative breast cancer, using breast cancer samples from two large international studies, The Cancer Genome Atlas and the Molecular Taxonomy of Breast Cancer International Consortium.
To move beyond subtype for identifying exactly which patients might develop resistance, they first sorted the most invasive tumor cells in frozen tissue using a molecular probe that was able to distinguish tumor from adjacent non-malignant cells or tissue by binding to an epigenetic regulator, or a histone methylase, called G9a. This enzyme has been reported by other scientists to be abnormally upregulated in many cancer types, including breast cancer.
They then identified select proteins that were working with G9a as partners-in-crime, and worked backwards from there to identify the genetic abnormalities linked to those partner proteins in the cancer cell. They found in many instances the genes for these interactor proteins were amplified in multiple copies, or abnormally overexpressed, rather than mutated.
"Nowadays, people think somatic mutations of select genes are the primary drivers of tumorigenesis," Chen said. "We didn't see many mutations on our identified driver genes. We actually found the genes encoding those interactors have a high frequency amplification in breast cancer patients with poor prognosis."
They then used this information to generate sets of genes that encoded these "interactor proteins," and identified those linked to poor prognosis in patients. Looking ahead, Chen and his colleagues plan to determine the specificity and sensitivity of multi-omic aberrations of particular interactor gene sets as new systems biomarkers to predict cancer patient prognosis.
More information: John A. Wrobel et al. Multi-omic Dissection of Oncogenically Active Epiproteomes Identifies Drivers of Proliferative and Invasive Breast Tumors, iScience (2019). DOI: 10.1016/j.isci.2019.07.001