Steven J. Fliesler, PhD, SUNY Distinguished Professor and the Meyer H. Riwchun Endowed Chair Professor of Ophthalmology, Jacobs School of Medicine and Biomedical Sciences. Credit: University at Buffalo
A series of papers published recently by two teams of vision researchers at the University at Buffalo and the University of Alabama at Birmingham (UAB) is revealing important new information about the possible cause of a condition that underlies a rare form of irreversible blindness in young children.
Retinitis pigmentosa (RP) is the label given to a family of disorders characterized by the degeneration and eventual death of the eye's photoreceptor cells, which absorb and convert light into electrical signals in the retina. Symptoms begin in childhood with night blindness and loss of peripheral vision, and the disease typically results in progressive vision loss and, ultimately, blindness.
The UB team is led by Steven J. Fliesler, Ph.D., SUNY Distinguished Professor and the Meyer H. Riwchun Endowed Chair Professor of Ophthalmology in the Jacobs School of Medicine and Biomedical Sciences at UB. The UAB team is led by Steven J. Pittler, Ph.D., professor and director of the Vision Science Research Center at the School of Optometry and Vision Science at UAB.
"RP is caused by a multitude of disparate genetic mutations," explained Fliesler. "However, one form known as RP59 has been classified as a congenital disorder of glycosylation (CDG). The process of glycosylation involves the addition of sugars to protein molecules to make glycoproteins, which are required for cellular development, structural integrity, and viability."
But the researchers' findings published this month in a paper in iScience, and two related papers published recently in the journal Cells, demonstrate that this may not be the case.
Genetic mutation
Although the more common forms of RP have been known for many decades, RP59 was only first described in 2011, in patients of Ashkenazi Jewish heritage. Those patients have a genetic defect involving mutations in the gene that encodes DHDDS, a key enzyme required for synthesizing an essential lipid-like molecule called dolichol. These patients manifest RP-like symptoms.
Up until then, Fliesler explained, it was neither known nor imagined that defects in the biochemical pathway that synthesizes dolichol could cause the disorder.
In 2018, the National Eye Institute of the National Institutes of Health awarded Fliesler and Pittler a $2.2 million grant to study RP59 with the hope of eventually developing a gene therapy to treat or cure the condition.
The aim of the grant, which supports the current work, was to study the molecular mechanism underlying RP59 pathology by deleting the DHDDS gene selectively in specific cell types in the retina and examining the consequences to retinal structure and function, Fliesler explained.
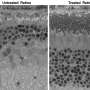
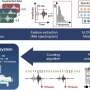
"The study described in the iScience paper involved targeted deletion in mouse retinal rod photoreceptor cells of the genes that encode a critical enzyme (DHDDS) responsible for making dolichol and its derivatives," said Fliesler. "Those mice exhibited a profound retinal degeneration with complete loss of rod photoreceptors by 6 postnatal weeks of age, which is among the most rapid photoreceptor degenerations ever observed.
"Although we predicted the retinal degeneration, we expected that protein glycosylation in the photoreceptor cells would be severely compromised, since that process requires dolichol; yet, it was not," said Fliesler. "That was a tremendous surprise.
"We have not yet discovered why this happens, or what is the actual underlying mechanism of the very rapid photoreceptor degeneration observed in our novel mouse model," said Fliesler. "Without knowing the underlying mechanism of RP59, it would be more difficult to develop targeted therapies to prevent, retard, or cure the disease. Our results, in combination with results obtained from two other recently published studies from our group, in collaboration with Dr. Pittler's lab at UAB, challenge the notion that RP59 is a congenital disorder of glycosylation."
The work is related to two previous papers, in which Fliesler and members of his UB lab are co-authors with the lead investigators at UAB. Published in Cells, the first study suggests that retinal pigment epithelium pathology may be a significant contributor to the retinal degeneration observed in humans with RP59 mutations.
More recently, the UAB and UB teams collaborated on a companion study, also reported in Cells, that showed that a "knock-in" mouse model (where an endogenous gene or parts of a gene have been replaced with exogenous genetic material) harboring the same mutation as found in human RP59 patients not only showed no defect in glycosylation, but failed to undergo retinal degeneration, and exhibited only very subtle physiological defects. These findings again point to a more complex mechanism for RP59 than originally thought, Fliesler noted.
More information: Sriganesh Ramachandra Rao et al. Retinal Degeneration Caused by Rod-Specific Dhdds Ablation Occurs without Concomitant Inhibition of Protein N-Glycosylation, iScience (2020). DOI: 10.1016/j.isci.2020.101198
Sriganesh Ramachandra Rao et al. Lack of Overt Retinal Degeneration in a K42E Dhdds Knock-In Mouse Model of RP59, Cells (2020). DOI: 10.3390/cells9040896
Journal information: iScience
Provided by University at Buffalo