Alteration of calcium channel signaling may explain the mechanism of autism spectrum disorder

Autism spectrum disorder (ASD) is a heterogeneous disorder initiated early in development and characterized by abnormal social communication. Accumulating evidence supports the idea that specific mutations affect regulatory proteins that control arrays of cellular pathways.
A special case of autism is known as the Timothy Syndrome (TS) caused by a point mutation in the alternatively spliced exon 8A of the calcium channel Cav1.2. TS is a multisystem disorder characterized also by cardiac dysfunction, causing sudden death from cardiac arrhythmias.
There are two Timothy mutations, G406R and G402S, that occur at the calcium channel Cav1.2. These two mutations modify the inactivation kinetics of the channel and by reducing voltage-dependent inactivation, causing an abnormal calcium overload leading to heart problems such as prolonged QT interval and cardiac arrhythmia. Surprisingly, only the G406R mutation is associated with ASD in four out of five patients carrying the mutation, while the G402S mutation fails to express the autistic phenotype.
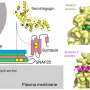
The study sought to answer the question: How does the G406R single point mutation of the Cav1.2 channel affect cellular processes that could lead to a multifactorial disease such as autism? It is known that Cav1.2 and other calcium channels induce gene activation, which is responsible for long-term processes, such as neurodevelopmental disorders, cognitive setbacks, and psychiatric disorders including schizophrenia, bipolar disorder and autism.
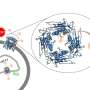
The researchers, led by Professor Daphne Atlas at the Hebrew University of Jerusalem's Alexander Silberman Institute of Life Sciences, found that both the Cav1.2 Timothy channel mutants G406R and G402S, activate gene programs (transcriptional activity) via the Ras/ERK/CREB cellular pathway, similar to the native (non-mutated) calcium channel Cav1.2. "We were surprised and thrilled to discover that the autistic mutant G406R exhibits a constitutive (spontaneous) transcriptional activation and the G402S mutation does not. This difference might clarify a mechanism that could explain why G406R mutation confers autism whereas G402S does not," shared Atlas.
It is known that in addition to modifying channel inactivation, the two variants of the Timothy channel differ in their activation kinetics. The G406R mutation causes specific gain-of-function changes in Cav1.2 channel gating, exhibiting a "leftward shift" of voltage-dependent activation and G402S does not. This leftward shift facilitates channel activity even at resting potentials, which means that the channel is spontaneously active, as opposed to the G402S mutant.
The research findings of facilitated spontaneous activity of the G406R channel correlates with a constitutive gene activation. This uncontrolled spontaneous gene activation, imposed by a leftward voltage-shift in the activation kinetics of the channel, implies a mechanism of conferring autism. The induction of uncontrolled long-term dysregulations such as autism by facilitated spontaneous activity of the channel and subsequently spontaneous gene activation can be compared to a dripping faucet.
These results were further supported through screening the literature for channel mutants that are associated with long-term abnormalities. All mutated channels that display a leftward-shift in channel activation are associated with long-term dysregulations. These findings imply that channel mutants that exhibit a facilitated channel activity at rest—with no stimulation—imposed by a negative shift in channel gating, are likely to exhibit a spontaneous and uncontrolled gene activation, similar to the Timothy mutant.
"Further studies are required to establish whether the uncontrolled activity of the channel at rest, which is associated with uncontrolled activation of gene programs in Timothy G406R mutant, is the underlying a mechanism by which other mutated channels confer a high risk for neurodevelopmental disorders in humans," explained Atlas.
Based on altering calcium channel kinetics and gene activation exhibited by the Timothy mutant, these results provide insight into the cellular mechanism that allows predicting disease risk, and genetic diagnosis of individuals with neurodevelopmental disorders.
The study was published in Progress in Neurobiology. It is part of a Ph.D. thesis by Evrim Servili and was done in collaboration with fellow HU colleagues, Drs. Michael Trus, Eilon Sherman and Julia Sajman.
More information: Evrim Servili et al. Elevated basal transcription can underlie timothy channel association with autism related disorders, Progress in Neurobiology (2020). DOI: 10.1016/j.pneurobio.2020.101820