A study of 181 patients at 16 sites across the country who test negative for two antibodies long known to cause muscle-weakening myasthenia gravis, found that about 15% test positive for one of two newly discovered antibodies that also attack the point of communication between nerves and muscle.
About 13% tested positive for antibodies to both LRP4 and agrin, proteins critical to healthy nerve-muscle communication, investigators report in the journal Muscle & Nerve about the largest group of patients examined for the new antibodies.
Those who tested positive for antibodies to LRP4, agrin or both, tended to be sicker when diagnosed and have a more severe disease course, investigators at Case Western Reserve University School of Medicine and the Medical College of Georgia at Augusta University report.
The investigators hope that learning more about antibodies that cause the disease will lead to treatments that target specific antibodies, which should reduce side effects and further improve efficacy.
Patients in the new study had no evidence of long-established antibodies for the receptor for acetylcholine, a chemical released by neurons, which activates muscle cell receptors, or MuSK, an enzyme that supports the clustering of these receptors on the surface of muscle cells and is essential to maintain the function of acetylcholine receptors.
Rather patients recruited for the study had been termed "double negative." Now the investigators say the roughly 85% of study participants who also tested negative for LRP4 and agrin are more accurately termed "quad negative."
Despite increased disease severity, better than 80% of patients with LRP4 and agrin antibodies had their disease moderated by standard therapy that includes a drug that increases acetylcholine levels by decreasing its breakdown as well as immunosuppressive drugs that reduce the body's overall production of antibodies.
All four now-established antibodies that cause myasthenia gravis fit together in a puzzle with the neuromuscular junction as their target, says Dr. Michael H. Rivner, MCG neurologist specializing in neuromuscular disease. "They are all functionally related to the neuromuscular junction even though they do different things," says Rivner, who directs the Electrodiagnostic Medicine Laboratory at MCG's adult teaching hospital AU Medical Center.
He and Dr. Lin Mei, chair of the Department of Neurosciences and Allen C. Holmes Professor of Neurological Diseases at Case Western Reserve University School of Medicine, are long-time collaborators and co-corresponding authors of the new study.
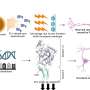
Mei's lab discovered the newer targets, reporting LRP4 antibodies as a new cause of myasthenia gravis in 2013 and agrin antibodies as associated with the disease the next year. Those findings filled important knowledge gaps in how the brain and muscle connect during development and continue to interact lifelong.
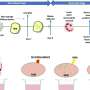
For our muscles to contract, they have to get a message from our nerves at the neuromuscular junction, which becomes the target of antibodies in myasthenia gravis. During development, agrin is released by motor neurons to direct construction of the synapse where nerves and muscles connect, then binds to the LRP4 receptor to activate MuSK, an enzyme on the muscle cell surface, which enables a clustering of receptors that are then activated by acetylcholine. While agrin and MuSK work together, they don't directly communicate, which is where LRP4 comes in. Mei's lab found that agrin communicates with LRP4 on the muscle cell surface, then recruits MuSK to join the conversation. LRP4 and MuSK become major components of the receptor needed for the muscle cell to receive the message agrin is sending.
LRP4 and agrin antibodies have been found in patients, and shown to produce myasthenia gravis in laboratory animals but there has been limited published data on their effect in humans.
Previously, about 10% of patients had no evidence of the two other antibodies already known to cause the disease. But they did have classic clinical symptoms such as general fatigue, blurred vision, drooping eyelids, generalized muscle weakness that gets worse with muscle use and, at its most aggressive, problems breathing, swallowing, even talking. When most patients first come to the doctor, it's because they are having problems with their droopy eyes and double vision, Rivner says, as the small muscle fibers and motor units that control them are ready targets for the antibodies.
In the newly published findings, about 70% of patients with the antibodies—compared with nearly 43% without them—had generalized symptoms of muscle weakness when they first sought medical care for those symptoms an average of 11 years ago.
Those with the antibodies also had a significantly higher severity of disease, including more eye problems, based on the Myasthenia Gravis Foundation of America's scale, with the state of disease classified as moderate to severe for 75% of the antibody positive patients compared with about 40% of the antibody negative individuals.
At their sickest, about half the patients with the antibodies had a mix of symptoms including difficulty with swallowing and speech, moving their arms and legs and breathing in additon to eye problems. "They started out sicker and became sicker," Rivner says. Twice as many patients in the antibody positive group needed mechanical ventilation support at some point, 11% versus 5% in the antibody negative group.
Regardless of their antibody status, patients were treated with standard therapies for myasthenia gravis including the drug pyridostigmine to improve muscle strength by blocking the breakdown of acetylcholine, plasma exchange to help clear circulating antibodies, and immunosuppressive drugs like prednisone. And most responded well to therapy, the investigators say.
The fact that patients respond to immunosuppressive drugs indicates they are making antibodies, Rivner says, which likely includes the "quad negative" patients. Immunotherapy helps diminish the immune system's attack on the neuromuscular junction, which can enable recovery of this communication point between nerves and muscle and help restore function to patients.
Next steps include analyzing five-year data on an even larger number of patients. They would also like to compare disease severity to patients with the longer-established antibodies.
About 80% of patients with myasthenia gravis have antibodies to the acetylcholine receptor and about 10% have antibodies to MuSK. The investigators chose to look at patients categorized as "double negative" to ensure that those antibodies were not a factor in their disease, Rivner says. However, he suspects many patients with those longer-established antibodies also have one or both of the newer ones.
More information: Michael H. Rivner et al. Clinical features of LRP4 /agrin‐antibody–positive myasthenia gravis: A multicenter study, Muscle & Nerve (2020). DOI: 10.1002/mus.26985
Provided by Medical College of Georgia at Augusta University