Credit: Jeffrey Decoster
"Know your enemy," Sun Tzu, the great sage of war, wrote some 2,500 years ago. Today, as COVID-19 spreads around the globe, the greatest army of medical scientists ever assembled is bent on learning all it can, as fast as it can, about SARS-CoV-2, the virus behind the pandemic.
Here's a primer on viruses in general and SARS-CoV-2 in particular. As researchers learn more and more about the novel coronavirus that causes COVID-19, this knowledge—gathered through unmatched levels of scientific cooperation—is being turned against the virus in real time.
Not that this will be a simple pursuit. Compared with a lab dish, living people are complicated. The cells in that dish aren't the same as the cells in living tissues affected by SARS-CoV-2. Plus, the environment surrounding, say, a lung cell in a person's body is different from the one in a culture dish. And then there's this thing called "side effects." You don't see those in a dish. But you may in a COVID-19 patient.
What, exactly, is a virus, anyway?
Viruses are easily the most abundant life form on Earth, if you accept the proposition that they're alive. Try multiplying a billion by a billion, then multiplying that by 10 trillion. That—10 to the 31st power—is the mind-numbing estimate of how many individual viral particles populate the planet.
Is a virus a living thing? Maybe. Sometimes. It depends on location. "Outside of a cell, a viral particle is inert," virologist Jan Carette, Ph.D., associate professor of microbiology and immunology, told me. On its own, it can't reproduce itself or, for that matter, produce anything at all. It's the ultimate parasite.
Or, you could say more charitably, it's very efficient. Viruses travel light, packing only the baggage they absolutely need to hack into a cell, commandeer its molecular machinery, multiply and make an escape.
There are exceptions to nearly every rule, but viruses do have things in common, said Carette.
A virus's travel kit always includes its genome—its collection of genes, that is—and a surrounding protein shell, or capsid, which keeps the viral genome safe, helps the virus latch onto cells and climb inside, and, on occasion, abets a getaway by its offspring. The capsid consists of identical protein subunits whose shapes and properties determine the capsid's structure and function.
Some viruses also wear greasy overcoats, called envelopes, made from stolen shreds of the membranes of the last cell they infected. Coronaviruses have envelopes, as do influenza and hepatitis C viruses, herpesviruses and HIV. Rhinoviruses, which are responsible for most common colds, and polioviruses don't. Enveloped viruses particularly despise soap because it disrupts greasy membranes. Soap and water are to these viruses what exhaling garlic is to a vampire, which is why washing your hands works wonders.
How do viruses enter cells, replicate and head for the exits?
For a virus to spread, it must first find a way into a cell. But, said Carette, "penetrating a cell's perimeter isn't easy." The outer membranes of cells are normally tough to get into without some kind of special pass. Viruses have ways of tricking cells into letting them in, though. Typically, a portion of the virus's cloak will have a strong affinity to bind with one or another protein that dots the surfaces of one or another cell type. The binding of the virus with that cell-surface protein serves as an admission ticket, easing the virus's invasion of the cell.
The viral genome, like ours, is an instruction kit for the production of proteins the organism needs. This genome can be made up of either DNA, as is the case with all creatures except for certain viruses, or DNA's close chemical relative RNA, which is much more flexible and somewhat less stable. SARS-CoV-2's genome is made of RNA, as are the genomes of most mammal-infecting viruses.
In addition to the gene coding for its capsid protein, every virus needs another gene for its own version of an enzyme known as a polymerase. Inside the cell, viral polymerases generate numerous copies of the invader's genes, from whose instructions the cell's obedient molecular assembly line produces capsid subunits and other viral proteins. Among these can be proteins capable of co-opting the cellular machinery to help viruses replicate and escape, or of tweaking the virus's own genome—or ours. Depending on the type of virus, the genome can contain as few as two genes—one for the protein from which the capsid is built, the other for the polymerase—or as many as hundreds.
Capsids self-assemble from their subunits, often with help from proteins originally made by the cell for other purposes, but co-opted by the virus. Fresh copies of the viral genome are packaged inside newly made capsids for export.
Often, the virus's plentiful progeny punish the good deed of the cell that produced them by lysing it—punching holes in its outer membrane, busting out of it and destroying the cell in the process. But enveloped viruses can escape by an alternative process called budding, whereby they wrap themselves in a piece of membrane from the infected cell and diffuse through the cell's outer membrane without structurally damaging it. Even then, the cell, having birthed myriad baby viruses, is often left fatally weakened.
Introducing the coronavirus, and how it latches on
Now we know how your average virus—an essentially inert particle on its own—manages to enter cells, hijack their molecular machinery, make copies of itself and move on out to infect again.
That just scratches the surface. Of the millions of different viral species identified so far, only about 5,000 have been characterized in detail. Viruses come in many shapes and sizes—although they're all small—and infect everything, including plants and bacteria. None of them works in precisely the same way.
So what about coronaviruses?
Enveloped viruses tend to be less hardy when they're outside of cells because their envelopes are vulnerable to degradation by heat, humidity and the ultraviolet component of sunlight.
This should be good news for us when it comes to coronaviruses. However, the bad news is that the coronavirus can be quite stable outside of cells because its spikes, protruding like needles from a pincushion, shield it from direct contact, enabling it to survive on surfaces for relatively long periods. (Still, soap or alcohol-based hand sanitizers do a good job of disabling it.)
As mentioned earlier, viruses use proteins that are sitting on cells' surfaces as docking stations. Coronaviruses' attachment-enabling counterpart proteins are those same spikes. But not all coronavirus spikes are alike. Relatively benign coronavirus variants, which at their worst might cause a scratchy throat and sniffles, attach to cells in the upper respiratory tract—the nasal cavities and throat. The viral variant that's driving today's pandemic is dangerous because its spike proteins can latch onto cells in the lower respiratory tract—the lung and bronchial cells—as well as cells in the lungs, heart, kidney, liver, brain, gut lining, stomach or blood vessels.
Antibody treatments could block binding
In a successful response to SARS-CoV-2 infection, the immune system manufactures a potpourri of specialized proteins called antibodies that glom on to the virus in various places, sometimes blocking its attachment to the cell-surface protein it's trying to hook onto. Stanford is participating in a clinical trial, sponsored by the National Institutes of Health, to see if antibody-rich plasma (the cell-free part of blood) from recovered COVID-19 patients (who no longer need these antibodies) can mitigate symptoms in patients with mild illness and prevent its progression from mild to severe.
So-called monoclonal antibodies are to the antibodies in convalescent plasma what a laser is to an incandescent light bulb. Biotechnologists have learned how to identify antibody variants that excel at clinging to specific spots on SARS-CoV-2's spike protein, thus thwarting the binding of the virus to our cells—and they can produce just those variants in bulk. Stanford is launching a Phase 2 clinical trial of a monoclonal antibody for treating COVID-19 patients.
A worry: Viral mutation rates are much higher than bacterial rates, which dwarf those of our sperm and egg cells. RNA viruses, including the coronavirus, mutate even more easily than DNA viruses do: Their polymerases (those genome-copying enzymes mentioned earlier) are typically less precise than those of DNA viruses, and RNA itself is inherently less stable than DNA. So viruses, and particularly RNA viruses, easily develop resistance to our immune system's attempts to find and foil them.
The Stanford studies may help reveal whether the precision-targeted "laser" or kitchen-sink "lightbulb" approach works best.
The virus breaks into a cell
Assistant professor of chemical engineering and subcellular-compartment spelunker Monther Abu-Remaileh, Ph.D., described two key ways the coronavirus breaks into a cell and seeks comfort there, and how it might be possible to bar one of those entry routes with the right kind of drug.
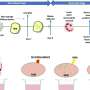
Here's one way: Once the coronavirus locks on to a cell, its greasy envelope comes into contact with the cell's equally greasy outer membrane. Grease loves grease. The viral envelope and cell membrane fuse, and the viral contents dump into the cell.
The other way is more complicated. The viral attachment can set off a process in which the area on the cell's outer membrane nearest the spot where the contact has been made caves in—with the virus (happily) trapped inside—until it gets completely pinched off, forming an inbound, membrane-coated, liquid-centered capsule called an endosome inside the cell. (To visualize this, imagine yourself with a wad of bubble gum in your mouth, blowing an internal bubble by inhaling, and then swallowing it. In this analogy, you're the cell and all your skin, beginning with your lips, constitutes the cell's outer membrane.)
Enclosed in this endosome is the viral particle that set the process in motion. The little devil has just hooked itself a ride into the cell's inner sanctum. At this point, the viral particle consists of its envelope, its capsid and its enclosed genome—a blueprint for the more than two dozen proteins the virus needs and the invaded cell doesn't provide.
But the endosome doesn't remain an endosome indefinitely, Abu-Remaileh told me. Its mission is to become another entity, called a lysosome, or to fuse with an existing lysosome.
Lysosomes serve as cells' recycling factories, breaking down large biomolecules into their constituent building blocks for reuse. For this, they need an acidic environment, generated by protein pumps on their surface membranes that force protons into these vesicles.
The building internal acidity activates enzymes that chew up the cloistered coronavirus's spike proteins. That brings the virus's envelope in contact with the vesicle membrane and enables their fusion.
The viral genome gets squirted out into the greater expanse of the cell. There, the viral genome will find and commandeer the raw materials and molecular machinery required to carry out its genetic instructions. That machinery will furiously crank out viral proteins—including the customized polymerase SARS-CoV-2 needs to replicate its own genome. Copies of the genome and the virus's capsid proteins will be brought together and repackaged into viral progeny.
A pair of closely related drugs, chloroquine and hydroxychloroquine, have gotten tons of press but, so far, mostly disappointing results in clinical trials for treating COVID-19. Some researchers advocate using hydroxychloroquine, with the caveat that use should be early in the course of the disease.
In a lab dish, these drugs diffuse into cells, where they diminish acidity in endosomes and prevent it from building up in lysosomes. Without that requisite acidity, the viral-membrane spike proteins can't be chewed up and the viral envelope can't make contact with the membrane of an endosome or lysosome. The virus remains locked in a prison of its own device.
That's what happens in a dish, anyway. But only further clinical trials will tell how much that matters.
How the coronavirus reproduces
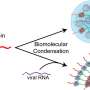
SARS-CoV-2 has entered the cell, either by fusion or by riding in like a Lilliputian aquanaut, stealthily stowed inside an endosome. If things go right, the virus fuses with the membrane of the surrounding endosome. The viral genome spills out into the (relatively) vast surrounding cellular ocean.
That lonely single strand of RNA that is the virus's genome has a big job to do—two, in fact, Judith Frydman, Ph.D., professor of biology and genetics, told me—in order to bootstrap itself into parenting a pack of progeny. It must replicate itself in entirety and in bulk, with each copy the potential seed of a new viral particle. And it must generate multiple partial copies of itself –— sawed-off sections that serve as instructions, telling the cell's protein-making machines, called ribosomes, how to manufacture the virus's more than two dozen proteins.
To do both things, the virus needs a special kind of polymerase, the protein that will function as a copying machine for the viral genome. Every living cell, including each of ours, uses polymerases to copy its DNA-based genome and to transcribe its contents (the genes) into RNA-based instructions that ribosomes can read.
The SARS-CoV-2 genome, unlike ours, is made of RNA, so it's already ribosome-friendly, but replicating itself means making RNA copies of RNA. Our cells never need to do this, and they lack polymerases that can.
SARS-CoV-2's genome, though, does carry a gene coding for an RNA-to-RNA polymerase. If that lone RNA strand can find and insert itself into a ribosome, the latter can translate the viral polymerase's genetic blueprint into a working protein. Fortunately for the virus, there can be as many as 10 million ribosomes in a single cell.
Once made, the viral polymerase cranks out not only multiple copies of the full-length viral genome—replication—but also individual viral genes or groups of them. These snippets can clamber aboard ribosomes and command them to produce the entire repertoire of all the proteins needed to assemble numerous new viral offspring.
These newly created proteins include, notably, more polymerase molecules. Each copy of the SARS-CoV-2 genome can be fed repeatedly through prolific polymerase molecules, generating myriad faithful reproductions of the initial strand.
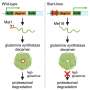
Well, mostly faithful. We all make mistakes, and the viral polymerase is no exception; actually it's pretty sloppy as polymerases go —– much more so than our own cells' polymerases, Carette and Frydman told me. So the copies of the initial strand—and their copies—are at risk of being riddled with copying errors, aka mutations.
However, coronavirus polymerases, including SARS-CoV-2's, come uniquely equipped with a sidekick "proofreader protein" that catches most of those errors. It chops out the wrongly inserted chemical component and gives the polymerase another, generally successful, stab at inserting the proper chemical unit into the growing RNA sequence.
Coronavirus birth control
The experimental drug remdesivir, approved for emergency use among hospitalized COVID-19 patients, directly targets RNA viruses' polymerases. Stanford participated in clinical trials leading to this injectable drug's approval. Initially developed for treating Ebola virus infection, it belongs to a class of drugs that work by posing as legitimate chemical building blocks of a DNA or RNA sequence. These poseurs get themselves stitched into the nascent strand and gum things up so badly that the polymerase stalls out or produces a defective product.
"Now, with the drug, the virus starts making a lot of rotten genomes that poison the viral replication process," said Frydman, the Donald Kennedy chair in the School of Humanities and Sciences.
Remdesivir has the virtue of not messing up our cells' own polymerases, said Robert Shafer, MD, professor of infectious disease, who maintains a continuously updated database of results from trials of drugs targeting SARS-CoV-2.
But while remdesivir's pretty good at faking out the viral polymerase's companion proofreader protein, it's far from perfect, Shafer said. Some intact viral genome copies still manage to get made, escape from the cell, and infect other cells—mission accomplished.
Using remdesivir in combination with some still-sought, as yet undiscovered drug that could block the proofreader might be a more surefire strategy than using remdesivir alone, Shafer said.
The final round in the cellular boxing ring
In addition to replicating its full-length genome, the virus has to make lots of proteins. And it knows just how. Those RNA snippets spun off by the viral polymerase are tailored to play by the cell's protein-making rules—well, up to a point. They fit into ribosomes exactly as do the cell's own strands of "messenger RNA" copied from the cell's genes by its own DNA-reading polymerases. So-called mRNAs are instructions for making proteins.
But there's a hitch: Among the proteins the virus forces ribosomes to manufacture are some that, once produced, bite the hand that fed them. Certain newly made viral proteins home to ribosomes in the act of reading one or another of the cell's mRNA strands, hook themselves onto the strand and stick stubbornly, stalling out the ribosome until the cell's mRNA strand falls apart.
The genomic RNA strands the virus generates, though, all have little blockades on their front ends that protect them from being snagged on the cell's ribosomes by the viral wrecking crew. The result: the cell's protein-making assembly line is overwhelmingly diverted to the production of viral proteins. That's a two-fer: It both increases viral-component production and stifles the infected cell's natural first line of defense.
Interferons as a potential treatment
Among the cell's stillborn proteins are molecules called interferons, which the cell ordinarlly makes when it senses it's been infected by a virus. Interferons have ways of monkeying with the viral polymerase's operations and squelching viral replication. In addition, when secreted from infected cells, interferons act as "call in the troops" distress signals that alert the body's immune system to the presence and location of the infected cell.
Instead, silence. Advantage: virus.
There are several different kinds of interferons. A clinical trial is underway at Stanford to determine whether a single injection of one of them, called interferon-lambda, can keep just-diagnosed, mildly symptomatic COVID-19 patients out of the hospital, speed recovery and reduce transmission to family members and the community.
One more thing about viruses
If you don't hate and respect viruses by now, maybe you haven't been paying attention. But there's more.
Viruses don't always kill the cells they take hostage. Some sew their genes into the genome of the cells they've invaded, and those insertions add up. Viral DNA sequences make up fully 8% of our genome—in contrast with the mere 1% that codes for the proteins of which we're largely made and that do most of the making.
"Our genome has been 'invaded' by previous encounters with retroviruses after infection of sperm or egg cells," Carette told me. "Through evolution, these retroviruses' genes have become inactive."
But, as always, there's an exception. As Carette told me: "An ancient viral gene has been repurposed to play an essential role in embryogenesis," the process by which an embryo forms and develops. The protein this gene encodes enables the fusion of two kinds of cells in the developing fetus's placenta, allowing nutrient and waste exchange between the developing embryo and the maternal blood supply.
Without them, that is, there'd be no us.
Provided by Stanford University