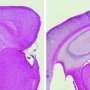
The endoplasmic reticulum strongly accumulates in KO synapses. Neurotransmitter-containing synaptic vesicles are shown in blue. Credit: Authors: Dmytro Puchkov, Marijn Kuijpers
It appears that autophagy protects neurons in the brain, but for different reasons than previously assumed, as researchers from the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) and Charité in Berlin have now shown. When the scientists used a genetic trick to switch off autophagy-mediated cellular waste disposal, instead of detecting protein deposits, as expected, they found elevated levels of the endoplasmic reticulum, a system composed of membrane sacs which acts, among other functions, as a calcium store. This leads to elevated neurotransmitter release and, ultimately, to fatal neuronal hyperexcitability. These fundamentally new findings have now been published in the prestigious journal Neuron.
Autophagy plays a key role in the maintenance of healthy cells, one example being the degradation and recycling of damaged protein molecules or entire organelles such as defective mitochondria by means of so-called autophagosomes. This cleaning mechanism is particularly important for neurons in the brain, which serve us throughout our lives, given that autophagy clears protein aggregates, such as those occurring in neurodegenerative diseases. The neuroprotective effects of autophagy have since been confirmed by numerous experiments in model organisms.
However, it is possible that the causes of this protective effect are different than previously assumed. By investigating the role of autophagy in the central nervous system of young, healthy mice, Professor Volker Haucke from the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) and his research group have now arrived at entirely new insights.
Using a genetic trick, the researchers first switched off an essential autophagy gene, and then used proteomics to analyze neuronal protein levels. Proteins previously hypothesized to be primarily degraded by autophagy were not enriched in the neurons at all—although this would have been expected, if their degradation occurred via autophagy.
"It came as a complete surprise to us," remarked Marijn Kuijpers, lead author of the study now published in Neuron. "But what surprised us even more was what we found in the neurons instead."
Largest intracellular calcium buffer no longer degraded
Instead of the expected autophagy substrates, the researchers discovered unusually high levels of the endoplasmic reticulum in the neuronal axons. One of the functions of these membrane sacs and tubules, which occur in all cells, is to provide a large intracellular store for calcium. The regulation of calcium is in turn fundamentally important for excitatory transmission in the central nervous system: When neurons communicate with each other, calcium channels open at synapses, leading to an influx of extracellular calcium into synapses and the release of neurotransmitters (neuronal messengers) from synaptic vesicles. Calcium can then either be pumped out of the neuron or enter the endoplasmic reticulum from where it can also be re-released, as required.
When autophagy was switched off, the calcium store of the endoplasmic reticulum turned out to be damaged. The researchers found that the calcium buffering function of the endoplasmic reticulum no longer worked properly, resulting in elevated calcium levels in axons and at synapses. This in turn boosted the release of the excitatory transmitter glutamate, which led to permanent neuronal hyperactivity.
Too much excitatory neurotransmitter release is the problem
"Until now, it was assumed that less autophagy meant the release of fewer transmitter molecules. We have now demonstrated the exact opposite," said postdoctoral fellow Marijn Kuijpers, commenting on the results of her study. "Too much, not too little neurotransmitter release is the problem. As a result, neurons become less plastic, and we suspect that they ultimately perish from hyperexcitability," added Charité's Professor Dietmar Schmitz, whose team contributed to the study.
Since the study was conducted with healthy neurons from young animals, it does not preclude additional functions of autophagy under pathological conditions, in Alzheimer's disease, for example. That said, the study is of tremendous importance for our fundamental understanding of the physiology of autophagy.
"All things considered, our discovery puts our understanding of autophagy in the central nervous system on a new basis," said group leader Professor Volker Haucke. This new information would explain, for example, why learning becomes more difficult as autophagy declines during aging. "It is not possible to ramp up a synapse that is already hyperactivated; it has reached its limit and can, thus, barely be plastically reinforced—a basic requirement for learning."
Using this new understanding to explore the key question of the trigger
The key question which regulatory mechanisms trigger autophagy in neurons remains open, however. While the availability of nutrients has a regulatory effect on other cells of the body—fasting has been shown to stimulate cellular waste disposal—no trigger for autophagy in the central nervous system is known so far.
"If we knew what produces more or less autophagy in neurons, we would be able at some point to therapeutically intervene," said Professor Haucke. "We are now keen to understand more about this fundamentally important problem, and our present study provides an excellent starting point for this endeavor."
More information: Kuijpers, M.; Kochlamazashvili, G., Stumpf, A., Puchkov, D, Swaminathan, A., Lucht, M., Krause, E., Maritzen, T., Schmitz, D., Haucke, V. Neuronal autophagy regulates presynaptic neurotransmission by controlling the axonal endoplasmic reticulum. Neuron (2020), Vol. 108, advance online, November 5 2020.
Journal information: Neuron
Provided by Forschungsverbund Berlin e.V. (FVB)