New statistical method eases data reproducibility crisis

A reproducibility crisis is ongoing in scientific research, where many studies may be difficult or impossible to replicate and thereby validate, especially when the study involves a very large sample size. For example, to evaluate the validity of a high-throughput genetic study's findings scientists must be able to replicate the study and achieve the same results. Now researchers at Penn State and the University of Minnesota have developed a statistical tool that can accurately estimate the replicability of a study, thus eliminating the need to duplicate the work and effectively mitigating the reproducibility crisis.
The team used its new method, which they describe in a paper publishing today in Nature Communications, to confirm the findings of a 2019 study on the genetic factors that contribute to smoking and drinking addiction but noted that it also can be applied to other genome-wide association studies—or studies that investigate the genetic underpinnings for diseases.
"While we applied the method to study smoking and drinking addiction-related outcomes, it could benefit other similar large-scale consortia studies, including current studies on the host genetic contribution to COVID-19 symptoms," said Dajiang Liu, associate professor of public health sciences and biochemistry and molecular biology, Penn State.
According to Liu, to detect patterns in genome-wide association studies it is important to obtain data from a large number of individuals. Scientists often acquire these data by combining many existing similarly designed studies, which is what Liu and his colleagues did for the 2019 smoking and drinking addiction study that ultimately comprised 1.2 million individuals.
"We worked really hard to collect all of the patient samples that we could manage," said Liu, noting that the data came from biobanks, epidemiology studies and direct-to-consumer genetic testing companies, such as 23andMe. However, he added, since the team used all of the available studies in its analysis, there were none leftover to use as comparisons for validation. "Our statistical method allows researchers to assess the replicability of genetic association signals without a replication dataset," he said. "It helps to maximize the power of genetic studies as no samples need to be reserved for replication; instead, all samples can be used for discoveries."
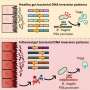
The team's method, which they call MAMBA (Meta-Analysis Model-Based Assessment of replicability), evaluates the strength and consistency of the associations between atypical bits of DNA, called single nucleotide polymorphisms (SNPs), and disease traits such as addiction. Specifically, MAMBA calculates the probability that if an experiment can be repeated with a different set of individuals, the relationships between the SNPs and those individuals' traits would be the same or similar as in the first experiment.
Qunhua Li, associate professor of statistics, Penn State, explained that MAMBA assigns a higher probability of replicability (PPR) for each SNP if the SNP is significantly associated with the trait being evaluated and if its estimated effect sizes are consistent across multiple studies.
"For example," said Li, "if the majority of participants who are addicted to smoking have a certain SNP that differs from non-addicted people, and if this SNP shows up across people in multiple smaller studies, then MAMBA will give it a higher PPR, which suggests that the SNP is probably important in addiction."
The researchers demonstrated the value of their method by applying it to Liu's 2019 study on smoking and drinking addiction. Among the 556 common and low-frequency SNP association signals, the team identified 529 with PPR greater than 99%. In an extended analysis of around 4,300 rare SNPs, the researchers identified 2,807 SNPs with PPR greater than 99%.
"Interestingly, we found that certain genes that are known to be responsible for lipid metabolism also influence smoking addiction," said Bibo Jiang, assistant professor of public health sciences, Penn State, noting that the phenomenon is known as pleiotropy—when a gene influences two seemingly irrelevant traits. "If we want to design medications that target those genes to help people stop smoking, we should be mindful of any underlying conditions related to lipid metabolism, such as high cholesterol, that they may have."
Liu noted that the method can be applied to genome-wide association studies focused on a wide variety of traits. "I think in the next decade or so, an essential focus of biology will be to interpret and make sense of those genome-wide association study discoveries and whether we can translate some of them into medications to facilitate personalized medicine," he said. "We are excited to be able to offer this statistical approach as a service to the research community."
More information: Nature Communications (2021). DOI: 10.1038/s41467-021-21226-z