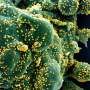
heavily infected with SARS-CoV-2 virus particles (red), isolated from a patient sample. Image captured at the NIAID Integrated Research Facility (IRF) in Fort Detrick, Maryland. Credit: NIAID")
Colorized scanning electron micrograph of a cell (blue) heavily infected with SARS-CoV-2 virus particles (red), isolated from a patient sample. Image captured at the NIAID Integrated Research Facility (IRF) in Fort Detrick, Maryland. Credit: NIAID
Using genome-wide association studies (GWAS) methodology to analyze whole-genome sequencing data of SARS-CoV-2 mutations and COVID-19 mortality data can identify highly pathogenic variants of the virus that should be flagged for containment, according to Harvard T.H. Chan School of Public Health and MIT researchers.
Using this biostatistical methodology, the researchers pinpointed a mutation in the variant known as P.1, or Gamma, as being linked to increased mortality and, potentially, greater transmissibility, higher infection rates, and increased pathogenicity before the P.1 variant had been identified.
The team's methodology is described online on June 23, 2021 in the journal Genetic Epidemiology.
"Based on our experience, GWAS methodology might provide suitable tools that could be used to analyze potential links between mutations at specific locations in viral genomes and disease outcome," said Christoph Lange, professor of biostatistics at Harvard Chan School and senior author of the paper. "This could enable better real-time detection of novel, deleterious variants/new viral strains in pandemics."
The first patients in Brazil with the P.1 variant were documented in January 2021 and within a few weeks the variant caused a spike in cases in Manaus, Brazil. The city had already been hard hit by the pandemic in May 2020, and researchers thought that the city's residents had achieved population immunity because so many people in the area had developed antibodies for the virus during that initial wave. Instead, P.1, which has several mutations in the spike protein the virus uses to attach to and invade a host cell, caused a second wave of infections and seemed to have higher transmissibility and be more likely to cause death than the earlier variants seen in the area.
In September 2020, several months before the first P.1 patient was documented, the Harvard Chan School and MIT team repurposed methodology used in GWAS, which are widely used to link certain genetic variations with specific diseases, to tease apart the relative pathogenicity of various SARS-CoV-2 mutations. The team looked for links between each mutation of the SARS-CoV-2 virus's single-stranded RNA and mortality in 7,548 COVID-19 patients. Data for the study came from the global initiative on sharing avian influenza data (GISAID) database, which contains the genetic sequence and related clinical and epidemiological data associated with SARS-CoV-2 and influenza viruses.
The researchers found one mutation—at locus 25,088bp in the virus's genome—that alters the spike protein and was linked to a significant increase in mortality in COVID-19 patients. The team flagged the variant with this mutation, which was later identified as part of P.1.
The team's biostatistical methodology should have broader applications beyond the P.1 variant and SARS-CoV-2, according to the researchers.
"We expect that this approach would work in similar scenarios involving other diseases, provided the quality of the data collected in public databases is sufficiently high," said Georg Hahn, research associate and instructor of biostatistics at Harvard Chan School and co-first author of the paper.
More information: "Genome-wide association analysis of COVID-19 mortality risk in SARS-CoV-2 genomes identifies mutation in the SARS-CoV-2 spike protein that colocalizes with P.1 of the Brazilian strain," Genetic Epidemiology, online June 23, 2021.
Provided by Harvard T.H. Chan School of Public Health