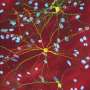
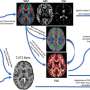
. The neuron in the center (yellow) contains an abnormal intracellular accumulation of huntingtin called an inclusion body (orange). Credit: Wikipedia/ Creative Commons Attribution 3.0 Unported license")
A montage of three images of single striatal neurons transfected with a disease-associated version of huntingtin, the protein that causes Huntington's disease. Nuclei of untransfected neurons are seen in the background (blue). The neuron in the center (yellow) contains an abnormal intracellular accumulation of huntingtin called an inclusion body (orange). Credit: Wikipedia/ Creative Commons Attribution 3.0 Unported license
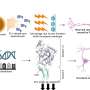
Mass spectrometry has emerged as an important analytical tool for gaining a better understanding of mechanisms underlying Huntington's disease (HD), alongside the increased availability of cell and animal models of the disease. This review, published in the Journal of Huntington's Disease, brings together and recaps data from major published mass spectrometry studies undertaken in HD research over the last 20 years, identifying important changes that occur in HD. The authors encourage researchers to make greater use of these studies to accelerate the development of new treatments.
HD is a rare neurodegenerative disorder caused by the aberrant expression of mutant Huntingtin (HTT) protein containing an expanded polyglutamine tract. Mass spectrometry is a technique that has existed for over a century to measure the mass-to-charge ratio of ions, however, proteins in complex mixtures such as tissues were not commonly analyzed until more modern ionization methods became available.
The authors have explained mass spectrometry in terms understandable for most biologists. Although these studies have yielded copious, useful data, researchers in the HD field have been unaware of just how many such studies have been performed.
"There are several advantages of using mass spectrometry," explained Kimberly B. Kegel-Gleason, Ph.D., MassGeneral Institute for Neurodegenerative Disease (MIND), Department of Neurology, Massachusetts General Hospital, Charlestown, MA, USA. "First, it is objective—scientists don't need to have a preformed idea or candidate proteins in mind, which frees the experiment of our preconceptions and allows us to identify changes that we might not have thought of. Second, recent advances in the hardware have made it easier to detect many proteins, including those at very low levels in a mixed sample. Finally, expanded gene databases provided by whole genome sequencing now allow accurate 'annotation' or identification of the proteins of interest."
HTT is now known to have a wide variety of post translational modifications (PTMs). In the early 2000s, mass spectrometry was used to identify Hap40 as part of protein complex with HTT. It was also used to investigate the difference in wild-type and HD proteomes (a proteome is the set of expressed proteins in a given type of cell, tissue, or organism). Following this study, several other investigators performed whole proteome studies in both human and mouse HD models. Many researchers have taken this a step further to investigate the HTT interactome (protein-protein interaction network) using mass spectrometry, and also examined HTT itself to identifying novel PTMs.
Notable among the studies reviewed are 15 proteome studies that sought to determine changes in expression levels (asking the question 'how much protein is there?') and five Interactome studies that looked at changes in how HTT interacts with other proteins (asking the question 'does HTT interact more or less with proteins?'). These studies compared brain tissue from animal models and autopsy tissue from patients with HD compared to controls. Importantly, three studies used cerebral spinal fluid from controls and patients with HD in an effort to identify biomarkers for disease progression. They also highlight four studies that identified posttranslational modifications (molecular markers) on the Huntingtin protein.
"Top-down mass spectrometry studies on the HTT protein are producing extremely interesting information about phosphorylation status and other modifications of HTT," noted co-author Connor Seeley, BS, Laboratory of Cellular Neurobiology, Department of Neurology, Massachusetts General Hospital, Charlestown, MA, USA. "Correlating these HTT proteoforms with functions should be fruitful in identifying mechanisms of pathology that can be targeted for intervention."
"To date, despite the large number of published works, there has not been a similar comprehensive review focused on results from HD mass spectrometry studies," commented Dr. Kegel-Gleason. "A substantial investment of research dollars was used to obtain these important data, yet often the greater community does not know how to interpret the findings or is unaware of how much data exist.
"We encourage researchers to use this resource of assembled studies to streamline their own research to accelerate our identification of treatments. Of special interest is validating biomarkers in cerebral spinal fluid (CSF). HD researchers should continue to mine data from these compiled works to support their findings. Finally, additional studies comparing control and HD CSF are warranted to discover and develop novel biomarkers for clinical applications."
HD is a fatal genetic neurodegenerative disease that causes the progressive breakdown of nerve cells in the brain. An estimated 250,000 people in the United States are either diagnosed with, or at risk for, the disease. Symptoms include personality changes, mood swings and depression, forgetfulness and impaired judgment, unsteady gait, and involuntary movements (chorea). Every child of an HD parent has a 50% chance of inheriting the gene. Patients usually survive 10 to 20 years after diagnosis.
More information: Connor Seeley et al, Taming the Huntington's Disease Proteome: What Have We Learned? Journal of Huntington's Disease (2021). DOI: 10.3233/JHD-200465
Journal information: Journal of Huntington\'s Disease
Provided by IOS Press