Researchers use prenatal editing in preclinical model to correct lysosomal storage disease

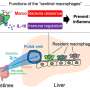
Adding to the growing body of literature demonstrating the feasibility of correcting lethal genetic diseases before birth, researchers at Children's Hospital of Philadelphia (CHOP) have used DNA base editing in a prenatal mouse model to correct a lysosomal storage disease known as Hurler syndrome. Using an adenine base editor delivered in an adeno-associated viral vector, the researchers corrected the single base mutation responsible for the condition, which begins before birth and affects multiple organs, with the potential to cause death in childhood if untreated.
The findings were published today in Nature Communications.
"This study shows that prenatal base editing for Hurler syndrome is feasible in a preclinical mouse model," said senior author William H. Peranteau, MD, an attending surgeon in the Division of General, Thoracic and Fetal Surgery at CHOP and Adzick-McCausland Distinguished Chair in Fetal and Pediatric Surgery. "In addition to showing the benefit of treating the disease before birth, we also showed some correction of the disease with base editing after birth, highlighting the promise of both pre- and postnatal base editing for Hurler syndrome."
Hurler syndrome, also known as Mucopolysaccharidosis Type I (MPS-IH), is a lysosomal storage disease that affects approximately 1 in 100,000 babies in the Western world and is commonly caused by a single DNA base mutation, where an adenine is present instead of a guanine. By 6 months of age, children can present with swelling of the liver and spleen, abdominal wall hernias, musculoskeletal abnormalities, retinal and neurocognitive degeneration, and cardiac disease. Untreated, patients can die of cardiorespiratory complications by 5 to 10 years of age. Even with treatment, patients experience complications, as current therapies have limited effectiveness, especially with delayed initiation.
Because the pathology of the disease begins before birth, the research team saw the syndrome as a candidate for prenatal treatment. With this goal, the researchers used CRISPR base editing, which requires only a single stranded DNA break and is thought to be more efficient and safer than other editing approaches, to convert the mutated adenine to guanine in the mouse model of MPS-IH.
The researchers used an adeno-associated virus serotype 9 (AAV9) viral vector to deliver the base editor to a fetal mouse model. They showed that prenatally treated mice demonstrated increased survival and improvement of metabolic, skeletal, and cardiac disease. Of note, the researchers observed corrected cells not only in the liver, but also in the heart, demonstrating the treatment was effective in multiple organs.
To assess the feasibility of the treatment after birth, the researchers tested the approach in 10-week-old MPS-IH mice and observed efficient on-target editing in the heart and liver, which was also associated with cardiac improvement. Whereas control models with the disease experienced cardiac decline between 4 and 6 months, sometimes resulting in death, postnatally treated mice demonstrated cardiac disease at 4 months, but the progression slowed between 4 and 6 months of age. Similar to prenatal treated mice, none of the mice treated after birth died by the study endpoint.
"Given the prenatal onset of disease, the potential for non-invasive prenatal diagnosis, and the progressive and morbid nature of the disease, Hurler syndrome and other lysosomal storage diseases represent attractive targets for treatment before birth," Peranteau said. "Although the safety of these approaches for mothers and fetuses still needs to be rigorously characterized prior to clinical translation, this proof-of-concept study offers hope for genetic diseases with limited postnatal treatments."
More information: Bose et al. "In utero split AAV9 adenine base editing corrects the multi-organ pathology in a lethal lysosomal storage disease," Nature Communications, online July 13, 2021, DOI: 10.1038/s41467-021-24443-8