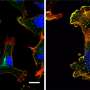
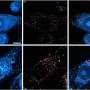
and after (right) treatment with an anticancer drug, which causes DNA breaks (green) in the cell nuclei")
Human lung cancer cells under the microscope, before (left) and after (right) treatment with an anticancer drug, which causes DNA breaks (green) in the cell nuclei
Desperate times call for desperate measures, says an ancient proverb, but whoever coined it surely could not have imagined that it would hold so true on such small scales. About half a century ago, scientists found that when bacteria are desperate to avoid a mortal threat, they activate a risky but effective genetic program called the SOS response.
Now, researchers at the Weizmann Institute of Science have discovered that cancer cells mobilize a similar SOS response when they are desperate to resist anticancer drugs. The researchers came up with an anti-SOS approach that blocked this mechanism in mice; it may one day be developed into an anticancer therapy for humans. Their study is published in Cancer Discovery
The team of scientists, headed by Prof. Yosef Yarden of Weizmann's Immunology and Regenerative Biology Department, investigated a puzzling subtype of lung cancer that affects nonsmokers. These cancers are peculiar not just because tobacco smoke is not involved but because their incidence varies greatly by geographical region. For example, they account for about 12% of all lung cancers in Israel and the United States, but for more than 35% in East Asia
In their initial stages, these cancers can be effectively treated. Their cells have mutations in the EGFR gene that are successfully targeted by a number of drugs. But after approximately a year, the drugs stop working because the tumors develop resistance: Secondary mutations enable them to evade the medications. The patients are usually treated with a second and later a third generation of drugs, but after each round, their tumors eventually become resistant to the drugs.
"We wanted to understand the underlying mechanisms of this resistance so that we could learn how to prevent it," Yarden says.
He drew inspiration from bacteriology, or more specifically, from studies of the SOS response in bacteria. This emergency system, first described by Croatian biologist Miroslav Radman in 1975, prompts the bacteria to generate large numbers of mutations throughout their DNA in response to a sudden life-threatening challenge such as exposure to a toxic chemical, an antibiotic or UV light.
"When the SOS response was discovered, scientists at first found it hard to believe," Yarden says. "Can bacteria really create so many mutations on purpose? After all, these mutations put organisms at an enormous risk. But the SOS response is only switched on when bacteria are on the verge of dying, so the risk pays off because at least some of the mutants turn out to be resistant to the threat."
This research direction proved extremely fruitful. Yarden and colleagues discovered that under the assault of anticancer drugs, lung tumors of nonsmokers develop resistance by means of a mechanism similar to the SOS response in bacteria. This conclusion emerged from experiments in tissue culture and in mice, led by Dr. Ashish Noronha, then a graduate student in Yarden's lab, in collaboration with a large team of scientists and physicians in Israel, Europe and the United States.
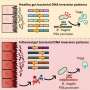
The researchers revealed how cancer's version of SOS works in all its molecular detail. When cancerous cells are killed in large numbers, the debris they leave behind triggers a mechanism in the survivors that swaps the enzymes responsible for copying DNA. Instead of high-fidelity enzymes that make faithful DNA copies, the new ones that take over, known as low-fidelity enzymes, are deliberately sloppy, introducing many new mutations during cell division. Yet another mechanism contributes to mutation by disrupting the supply of proper quantities of the four nucleotides, A, T, G and C, the building blocks needed for copying DNA. That mechanism creates an abnormal excess of one pair of nucleotides, A and G, over the other pair, which inevitably leads to errors in the copying process.
Most important, the researchers managed to identify the central regulator of the cancer's SOS response: a cellular surface receptor called AXL. It acts not only as a sensor that picks up signals from dying cancerous cells but also as a master switch that turns on the generation of mutations. And it's fast: Within one year, all cells in a tumor may harbor resistance-providing mutations.
Armed with this knowledge, the researchers started looking for ways of blocking cancer's SOS system. They found that by eliminating the AXL receptor in the tumor cells, they could prevent resistance from emerging. "This was the eureka moment—we knew we should look for a therapy that would disable this receptor," Yarden says.
This task, however, proved harder than expected. The researchers created an antibody that blocks AXL in mice, but it failed to produce sufficiently strong effects, whether given on its own or in combination with Tagrisso, a major drug used to treat lung cancers characterized by EGFR mutations. Only when the researchers gave mice a triple therapy—the anti-AXL antibody, Tagrisso and an additional anticancer drug, Erbitux, developed on the basis of research by Yarden and the late Prof. Michael Sela—did the tumors fail to develop resistance, and they were eliminated for good.
Producing effective humanized versions of the anti-AXL antibody and testing the triple therapy in clinical trials may take quite a few years. But in the future, the study's findings may lead to a promising new therapy for treating lung cancer in nonsmokers and perhaps other cancer subtypes that currently tend to develop resistance to existing medications.
More information: Ashish Noronha et al, AXL and error-prone DNA replication confer drug resistance and offer strategies to treat EGFR-mutant lung cancer, Cancer Discovery (2022). DOI: 10.1158/2159-8290.CD-22-0111
Journal information: Cancer Discovery
Provided by Weizmann Institute of Science