. From the paper: COVID-19 Infection and Transmission Includes Complex Sequence Diversity, PLOS Genetics, September 8, 2022. Credit: Ernest (Ricky) Chan")
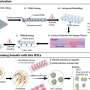
Whole genome sequencing of 140 COVID samples from infected individuals between April 2020 and August 2021 show infection signatures consistent with the major strain at the time represented in different color partitions. Each horizontal line illustrates a sequenced sample, and lineage specific positions are shown vertically. Gray shaded boxes represent mixed allele frequencies observed in our samples. Interestingly, we observe samples that represent mixed allele frequencies during the transition between B.1.1.7 and B.1.617.2 (purple partition). From the paper: COVID-19 Infection and Transmission Includes Complex Sequence Diversity, PLOS Genetics, September 8, 2022. Credit: Ernest (Ricky) Chan
Researchers at Case Western Reserve University found wide genetic variation in SARS-CoV-2 viruses among 360 patients whose viral infections were genetically sequenced, showing that all individual infections include multiple variants of the virus.
The researchers noted that reporting about the virus usually highlights a single dominant strain, which leads to under-reporting virus genetic variation and can have serious consequences in public-health planning and response.
"Our work brings attention to the complexity of infectious diseases that is often over-simplified when considering only the most abundant virus in an infection, and we demonstrate the importance of examining the variations that are historically considered noise," said Ernest (Ricky) Chan, director of the bioinformatics core with the Cleveland Institute for Computational Biology at the Case Western Reserve School of Medicine. "We see that genetic variants observed in low frequency in SARS-CoV-2 infections can be early indicators of new strains responsible for later transmission surges."
The paper, "COVID-19 Infection and Transmission Includes Complex Sequence Diversity," will be published on September 8, 2022 in PLOS Genetics.
The CWRU team performed full genome sequencing of SARS-CoV-2 viruses from 250 patients in Northeast Ohio and used similar data from another 110 patients with full genetic sequences of infecting viruses provided through international research collaborators.
These data were developed in the early days of the COVID-19 pandemic when the Alpha variant and then the Delta variant were of major concern. This work showed that mutations found in Omicron BA.1 and BA.2 were already present as relatively minor variations at least a year before Omicron and its many iterations became "variants of concern." Omicron and its own variants were central to a major COVID-19 resurgence last winter.
"Concentration on a majority consensus of virus variants within the global research community diverts attention from genetic variation that may contribute significantly to the continuing evolution of the COVID-19 pandemic," said Peter Zimmerman, a professor in the Department of Pathology at the School of Medicine. "Focus on majority variants is a critical first step in development of diagnostics, therapeutics and vaccines, however the research community needs to quantify and report out variation, so that the public health community and the general public are better prepared and nimble in response to the ever-evolving virus."
Much continues to be made to define and track the emergence of virus lineages across the ongoing evolution of SARS-CoV-2 around the world. In the interest of time, global researchers have been relying on tracking and reporting on relatively dominant variations. But the CWRU researchers noted that, given the multiple variations within single infections, it is important to report a more complete representation of the viral genetic sequences to understand how these genetic changes can spread and potentially interact with different categories of patient conditions, including evasion from eradication efforts.
More information: COVID-19 infection and transmission includes complex sequence diversity, PLoS Genetics (2022). journals.plos.org/plosgenetics … journal.pgen.1010200
Journal information: PLoS Genetics
Provided by Case Western Reserve University