A link between hypoxia and fetal hemoglobin provides hope for sickle cell disease
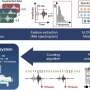
 enzymes and targeted for degradation by the VHL E3 ubiquitin ligase complex (not shown). The transcriptionally silent BGLT3, HBE, and γ-globin (HBG1 and HBG2) genes interact via chromatin looping, whereas the upstream LCR enhancer interacts with the active adult β-globin gene (HBB) to drive its transcription. At low oxygen levels, PHD enzymes are inactive and HIF1α accumulates and heterodimerizes with HIF1β. The HIF heterodimers bind two tandem HRE elements in BGLT3, leading to the recruitment of the transcriptional activators P300 and GATA1 (not shown), the acquisition of local enhancer functions, and the induction of the BGLT3 long noncoding RNA transcript (not shown). Activated BGLT3 unlocks the interaction with HBE, redirects the LCR to the γ-globin genes, and activates their transcription. The solid arrows indicate mRNA transcription. Yellow indicates transcriptionally active genes. Credit: Nature (2022). DOI: 10.1038/s41586-022-05312-w")
Scientists at St. Jude Children's Research Hospital have shown how a protein responsible for adapting to low oxygen conditions (hypoxia), causes increased expression of fetal hemoglobin (HbF) in adults. The finding has implications for treating sickle cell disease and beta-thalassemia, serious blood disorders that affect millions of individuals. The research was published today in Nature, with associated code published on GitHub.
Hemoglobin is like a protein sponge that soaks up oxygen and allows red blood cells to ferry it throughout the body. Adult hemoglobin contains four protein subunits—two beta-globin and two alpha-globin.
Mutations in beta-globin cause sickle cell disease and beta-thalassemia. But humans have another hemoglobin subunit gene (gamma-globin), which is expressed instead of beta-globin during fetal development. Gamma-globin combines with alpha-globin to form HbF. Normally around birth, gamma-globin expression is turned off and beta-globin is turned on, resulting in a switch from HbF to adult hemoglobin.
"We have known for many years that persistent HbF expression after birth can alleviate the symptoms of sickle cell disease and beta-thalassemia," said corresponding author Mitchell J. Weiss, M.D., Ph.D., St. Jude Hematology Department chair. "And very high HbF levels can cure these diseases, despite the defective beta-globin genes being present. Therefore, many laboratories are focused on understanding the perinatal switch from gamma- to beta-globin gene expression and figuring out new ways to reverse it with drugs or genetic therapies."
Restoring HbF production in adults
The St. Jude group discovered that hypoxia inducible factor 1 (HIF1) directly promotes transcription of the gamma-globin gene to enhance HbF production. HIF1 is an important component of cells' ability to sense and adapt to hypoxic conditions. In low oxygen conditions, HIF1 accumulates in many tissues and activates hundreds of genes, including HbF in red blood cells.
First author Ruopeng Feng, Ph.D., a scientist in the Weiss lab, showed that a drug that activates part of the cellular hypoxia response inhibits sickling of red blood cells derived from adults with sickle cell disease. The drug, a proline hydroxylase inhibitor, caused HIF1 to accumulate, bind a DNA regulatory region near the gamma globin gene, activating its transcription to produce HbF and inhibit cell 'sickling.'
Proline hydroxylase inhibitors are currently in late stage clinical development for the treatment of anemia associated with chronic kidney disease. These drugs work by stabilizing HIF proteins to stimulate the production of erythropoietin, a hormone that drives red blood cell production.
"Our findings indicate that proline hydroxylase inhibitors might be useful for treatment of sickle cell disease or beta-thalassemia, where turning on HbF production has therapeutic benefits," Weiss said. "Approximately 20% of adult sickle cell disease patients develop kidney failure with related anemia. Proline hydroxylase inhibitors might serve a dual purpose in these individuals, by stimulating the production of both erythropoietin and HbF."
Tying the hypoxia response to HbF
The Nobel Prize in Physiology or Medicine was awarded to the discovery of the HIF pathway in 2019. The current study, led by Weiss' group, establishes a direct connection between this hypoxia adaptation and HbF expression. This connection explains longstanding clinical observations that HbF is induced during accelerated production of red blood cells after exposure to hypoxia or in some forms of anemia, conditions termed "stress erythropoiesis."
"Identification of gamma-globin as a HIF target gene supports the notion that HbF evolved as a protective mechanism against hypoxia," Weiss said. "Studies of hemoglobin over more than 50 years have established many general principles in biology and medicine. It is exciting and gratifying that investigations into hemoglobin and globin gene expression continue to produce new, clinically relevant discoveries."
More information: Mitchell Weiss, Activation of γ-globin expression by hypoxia-inducible factor 1α, Nature (2022). DOI: 10.1038/s41586-022-05312-w
Code: github.com/YichaoOU/HemTools