New informatics software helps identify rare genetic variants

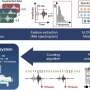
A team of researchers at Indiana University School of Medicine has developed specialized bioinformatics software designed to identify rare genetic variants in whole-genome sequencing studies. Zilin Li, Ph.D., assistant professor of biostatistics and health data science, was the first and co-corresponding author of the recent publication in Nature Methods which details the variant-Set Test for Association using Annotation infoRmation pipeline (or STAARpipeline) framework.
"Even though there are hundreds of millions of rare genetic variants, they have been challenging to study because there was no convenient, scalable and robust pipeline for comprehensive rare-variant analysis, which requires the evaluation of variant sets rather than single variants," Li said.
The STAARpipeline allows researchers to evaluate sets of rare, noncoding genetic variants, which will help enable genetic research. Noncoding genetic variants are parts of the genome that do not code for amino acids, the molecules that combine to form proteins. More than 98 percent of a person's DNA is noncoding.
"Rare variants are observed in 99% of the human genome and are a major source of the missing heritability of complex traits and diseases," Li said.
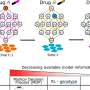
To use the STAARpipeline, researchers input genotype (genetic code) and phenotype (complex trait or disease code) data into the program. The software analyzes that data and identifies rare variants, grouping the variants into eight functional categories in the gene-centric analysis and into fixed-size sliding windows and newly proposed data-adaptive dynamic windows in the non-gene-centric analysis. The gene-centric analysis focuses on variants in or near genes, while the non-gene-centric analysis focuses on variants in the intergenic region, which is the stretch of DNA located between genes. The program then incorporates multiple variant functional annotations for each variant set to increase analysis power further and summarizes the results for the user.
The research team has already tested the STAARpipeline on large sample sizes, including 40,000 from the National Heart, Lung and Blood Institute (NHLBI) Trans-Omics Precision Medicine Program. During that analysis, STAARpipeline found 49 significant associations in gene-centric noncoding analysis, 35 of which were found based on six new proposed noncoding categories. In addition, data-adaptive size dynamic window analysis detected 43 non-overlapping significant associations in the noncoding genome, 19.4% more than the classical fixed-size sliding window procedure.
The STAARpipeline builds on STAAR, another program Li and his colleagues established, which is a genetic variant-set test for finding connections and associations by using annotation information.
"We believe the STAARpipeline can be expanded to analyze hundreds of millions of variants worth of whole genome sequencing data," Li said. "Since rare variants have been found in 99 percent of the human genome, this program addresses an important gap in informatic analysis."
More information: STAARpipeline: an all-in-one rare-variant tool for biobank-scale whole-genome sequencing data, Nature Methods (2022). DOI: 10.1038/s41592-022-01641-w