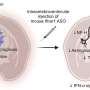
A schematic protocol of the MyoD1-induced differentiation for 10 days, with reference to our previous report. (b) Time-course of mRNA expressions of the DM1-related genes, DMPK and MBNL1 by RT-qPCR analyses on day 0, day 3 and day 10 of differentiation. The gene expressions are indicated by relative values to 414C2 control on day 0. The data represent the means of three independent experiments and were analyzed by one-way ANOVA followed by Tukey’s test (*P")
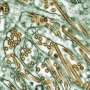
Nuclear MBNL1 aggregation in the myotubes differentiated from MyoD-DM1-hiPSCs. (a) A schematic protocol of the MyoD1-induced differentiation for 10 days, with reference to our previous report. (b) Time-course of mRNA expressions of the DM1-related genes, DMPK and MBNL1 by RT-qPCR analyses on day 0, day 3 and day 10 of differentiation. The gene expressions are indicated by relative values to 414C2 control on day 0. The data represent the means of three independent experiments and were analyzed by one-way ANOVA followed by Tukey’s test (*P < 0.05, **P < 0.01, vs. 414C2 control and ## P < 0.01, ### P < 0.001, vs. 409B2 control). (c) The representative immunohistochemistry images for MyHC and Hoechst on day 10. Scale bars, 100 μm. (d and e) Evaluation of nuclear MBNL1 aggregation on day 10. (d) Representative immunohistochemistry images after PFA or acetone-MeOH fixation for MBNL1 and Hoechst. Scale bars, 10 μm. (e) Quantitative analyses of the images of MBNL1 immunostaining after acetone-MeOH fixation using the ImageJ software. The nuclear regions were detected by Hoechst counterstaining. Data represent the means + SD of three independent experiments and were analyzed by one-way ANOVA followed by Tukey’s test (***P < 0.001). Credit: Scientific Reports (2023). DOI: 10.1038/s41598-022-26614-z
The Hidetoshi Sakurai Laboratory at CiRA has successfully reproduced the pathology of Myotonic Dystrophy type 1 (DM1) using skeletal muscle cells generated from patient-derived iPS cells and demonstrated their use for quantitative assessment of drug efficacy.
DM1 is considered the most frequent muscular dystrophy in adults, but there are currently no therapeutic treatments available. In order to discover and develop new therapeutic agents, there is an urgent need to devise an evaluation system using human cells that can faithfully recapitulate the pathology observed in DM1 patients.
DM1 is an inherited disease caused by the abnormal expansion of CTG sequence repeats in the DMPK (dystrophia myotonica-protein kinase) gene. Abnormal RNA generated from the CTG-expanded DMPK gene induces aggregation and consequently a loss of function of the splicing factor MBNL1 (muscleblind-like splicing regulator 1) in the nucleus. The resulting disruption of MBNL1-mediated gene splicing is believed to be the main mechanism of DM1 pathogenesis.
Using both direct and indirect differentiation protocols they devised previously, with or without forced MYOD1 expression, respectively, the research group was able to generate skeletal muscle cells using iPS cells derived from DM1 patients and replicate various DM1 cellular pathological changes such as the formation of MBNL1 protein aggregates and splicing defects of genes such as DMD, BIN1, and ATP2A1.
The group next used these differentiated cells to evaluate drug candidates for their abilities to reverse the observed cellular pathologies. Differentiated DM1 skeletal muscle cells were treated with CAG25, an antisense oligonucleotide previously shown to exhibit therapeutic effects against DM1 pathology in preclinical studies. Consistently, MBNL1 aggregation and splicing defects in the differentiated DM1 skeletal muscle cells were both rescued by CAG25 treatment, suggesting that these differentiated skeletal muscle cells can be used as a platform to quantitatively assess drug efficacy based on known DM1 pathology.
The study is published in the journal Scientific Reports.
This newly constructed drug evaluation system based on differentiated skeletal muscle cells produced from DM1 patient-derived iPS cells is expected to contribute to future discovery and development of effective therapeutic agents against DM1.
More information: Ryu Kawada et al, Establishment of quantitative and consistent in vitro skeletal muscle pathological models of myotonic dystrophy type 1 using patient-derived iPSCs, Scientific Reports (2023). DOI: 10.1038/s41598-022-26614-z
Journal information: Scientific Reports
Provided by Kyoto University