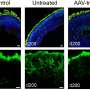
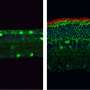
. Reserpine restores the balance between autophagy and the ubiquitin-proteasome system histone deacetylase 6 and improves primary cilium assembly. Credit: Holly Y. Chen, Ph.D., NEI")
In LCA10, CEP290 mutations lead to defects in the photoreceptor outer segment. Consequently, the building blocks of the primary cilium accumulate in the photoreceptors, which activates autophagy (shown left, untreated). Reserpine restores the balance between autophagy and the ubiquitin-proteasome system histone deacetylase 6 and improves primary cilium assembly. Credit: Holly Y. Chen, Ph.D., NEI
A National Institutes of Health team has identified a compound already approved by the U.S. Food and Drug Administration that keeps light-sensitive photoreceptors alive in three models of Leber congenital amaurosis type 10 (LCA 10), an inherited retinal ciliopathy disease that often results in severe visual impairment or blindness in early childhood.
LCA 10 is caused by mutations of the cilia-centrosomal gene (CEP290). Such mutations account for 20% to 25% of all LCA—more than any other gene. In addition to LCA, CEP290 mutations can cause multiple syndromic diseases involving a range of organ systems.
Using a mouse model of LCA10 and two types of lab-created tissues from stem cells known as organoids, the team screened more than 6,000 FDA-approved compounds to identify ones that promoted survival of photoreceptors, the types of cells that die in LCA, leading to vision loss. The high-throughput screening identified five potential drug candidates, including Reserpine, an old medication previously used to treat high blood pressure.
Observation of the LCA models treated with Reserpine shed light on the underlying biology of retinal ciliopathies, suggesting new targets for future exploration. Specifically, the models showed a dysregulation of autophagy, the process by which cells break down old or abnormal proteins, which in this case resulted in abnormal primary cilia, a microtubule organelle that protrudes from the surface of most cell types.
In LCA10, CEP290 gene mutations cause dysfunction of the primary cilium in retinal cells. Reserpine appeared to partially restore autophagy, resulting in improved primary cilium assembly.
Reserpine targets dysregulated intracellular signaling pathways downstream of the primary cilium. Such a treatment strategy could potentially address retinal ciliopathies caused by many of the more than 160 disease-causing genes, regardless of the specific gene involved. That's in contrast to gene therapy, which requires a very expensive and labor-intensive process to tailor an individual gene-based therapeutic approach for each mutation.
The findings are published in the journal eLife.
More information: Holly Y Chen et al, Reserpine maintains photoreceptor survival in retinal ciliopathy by resolving proteostasis imbalance and ciliogenesis defects, eLife (2023). DOI: 10.7554/eLife.83205
Journal information: eLife
Provided by National Institutes of Health