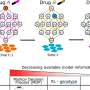
. DOI: 10.1016/j.cell.2023.02.023")
Credit: Cell (2023). DOI: 10.1016/j.cell.2023.02.023
Just like pruning a tree helps promote proper growth, the brain uses synaptic pruning to get rid of unnecessary connections between its cells. However, when this normal process, which occurs between early childhood and adulthood, doesn't stop properly, the brain loses too many connections, including important ones. Because of this excessive pruning, some brain cells die and others cause inflammation, leading to problems with movement, thinking and learning.
In a recently published pioneering study, a research team from the Research Institute of the McGill University Health Centre (RI-MUHC) and the Lady Davis Institute (LDI) at the Jewish General Hospital figured out how this faulty process occurs.
By examining the developmental consequences of three specific histone mutations (H3.3G34R, V and W), researchers led by Dr. Nada Jabado and Dr. Livia Garzia at the RI-MUHC, together with Dr. Claudia Kleinman at the LDI, unraveled a genetic mechanism that leads to severe neurodevelopmental syndromes. Specifically, they discovered how the brain gets damaged in diseases caused by these germline mutations—mutations that are present in reproductive cells and that become incorporated into every cell's DNA.
Their findings, published in the journal Cell, could not only help scientists find new ways to treat these diseases but also shed a light on the study of other neurological diseases that involve brain cell loss and inflammation, such as Alzheimer's, as well as disorders where excessive pruning is suspected, such as schizophrenia.
"Neurons cannot be replaced. Finding mechanisms that may affect them is important and opens the door to therapeutic interventions that are direly needed, both to restrain inflammation before neuronal loss becomes important; and to contain disease in the long term," says Dr. Jabado, a senior scientist in the Child Health and Human Development Program at the RI-MUHC and a professor in the Department of Pediatrics at McGill University.
"Neurodegeneration is one of the major issues we face as we age," adds Dr. Kleinman, an associate professor of Human Genetics at McGill University and principal investigator at the LDI. "Any new insight is welcome as the needs are huge, and our findings may shed another light on this devastating problem."
Persistent instead of transient pruning
The study shows that specific mutations in histone genes lead to decreased expression of "beacons" on specific areas of the chromatin (a mixture of DNA and proteins that form the chromosomes). These beacons usually recruit DNMT3A, an enzyme that can silence or activate genes depending on areas of deposition. Their decrease allows genes to keep producing proteins that contribute to the synaptic pruning process. Thus, the pruning that should have stopped continues, causing brain damage.
"We knew that severe neurodevelopmental syndromes, which are associated with a smaller brain, trouble walking, eating and speaking, as well as difficulty with learning, were caused by certain germline mutations in histones," explains Dr. Livia Garzia, a scientist in the Cancer Research Program at the RI-MUHC and assistant professor in the Department of Surgery at McGill University. "In this study, we demonstrated what happens when histones can't do their job properly and let the brain make more inflammatory proteins than needed, causing progressive neurodegeneration."
It all started with two patients
Dr. Jabado's team identified a patient with de novo (not inherited from his parents) germline mutation in H3.3G34R. The patient showed common characteristics with another patient having a mutation in H3.3G34V. Both patients had severe neurodevelopmental delay from birth, which worsened during postnatal development. At age 2, they also had severe growth delay and microcephaly.
"Both patients had other clinical symptoms, such as feeding difficulties, repetitive hand movements and drug-resistant epilepsy, and neither of them was able to walk or talk, which suggest that the H3.3G34R/V germline severely impaired their brain development and neurocognitive function," explains Dr. Jabado, who is also a pediatric hemato-oncologist at the Montreal Children's Hospital of the MUHC. "However, patient G34R showed a distinctive developmental regression, illustrated by loss of sitting ability, as well as decreased social interactivity."
Distinct observations for each mutation
To understand the developmental consequences of H3.3G34 germline mutations, the researchers engineered mice with G34R, V, and W mutations using embryo editing.
In the study article, the researchers report that the neurological impairments manifested in mice during adolescence and early adulthood and progressively worsened with age. They also noted that G34R, V and W mutations fundamentally affected brain development in surprisingly different ways. For instance:
- G34R mutation affected tissues of a different origin than G34W.
- G34R mice exhibited severe neurological alterations, while G34W mice had bladder, uretero-genital defects and morbid obesity.
- G34R mice, but not G34W, showed impaired motor functions.
- Ataxia, a condition that affects coordination, balance and speech due to poor muscle control, severely affected G34R mice and caused their death. It did affect G34V mice to a modest extent but was absent in G34W models.
- G34R mutation, and to a lesser extent G34V but not G34W, induced progressive microcephaly.
"In our investigation, we were able to detect remarkable developmental differences in H3.3G34R, V and W mutants, reminiscent of the specificity observed in patients with H3.3G34-mutant diseases," says Dr. Jabado. "We elucidated G34R-mediated epigenetic dysregulations in the developing brain, potentially underlying the neurological deficits that are unique to this mutation."
Getting the full story from gene to phenotype
The team of researchers is internationally known for their work on cancer and brain tumors, specifically on mutations in histone genes, and it is in that context that they were initially studying H3.3G34 mutations in the lab. At one point, they stumbled across mice who were showing a phenotype (a set of characteristics) of neurodegeneration for a specific mutant and uncovered patients with neurodegeneration who had similar symptoms.
"We could not let go based on how severe and compelling this phenotype was. In addition, the fact that this mutation may also occur in the germline, severely affecting children and negating their future, was a major incentive to move forward," explains Dr. Jabado.
Sima Khazei, a graduate student in Dr. Jabado's lab, was crucial in this development. With hard work, acumen and perseverance, she led the team as it tried to understand why they were seeing this phenotype. The answer came when everyone—Drs. Jabado, Garzia and Kleinman, as well the first co-authors Sima Khazaei, Carol CL Chen and Augusto Faria Andrade—each provided a piece of the puzzle, validating data that made this cascade of events real and potentially applicable elsewhere.
"We are proud of this work, not only because of the findings, but also because of the approach we used to unravel mechanisms of disease and the tools we have provided to the scientific community, both of which are valuable and could be applied to study several types of brain disorders," says Dr. Kleinman.
More information: Sima Khazaei et al, Single substitution in H3.3G34 alters DNMT3A recruitment to cause progressive neurodegeneration, Cell (2023). DOI: 10.1016/j.cell.2023.02.023
Journal information: Cell
Provided by McGill University Health Centre