This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Bold new therapy delivery method shows initial promise as treatment for Duchenne muscular dystrophy
. DOI: 10.1016/j.cell.2023.03.033")
Doug Millay, Ph.D., a scientist with the Division of Molecular Cardiovascular Biology at Cincinnati Children's has dedicated his career to revealing the most fundamental mechanisms of skeletal muscle development. He has been a leader in characterizing how two "fusogens" called Myomaker and Myomerger mediate the entry of stem cells into mature muscle cells to build the tissue that humans depend upon for movement, breathing, and survival.
Now, some of the basic discoveries made by Millay and colleagues are translating into a potential treatment for people living with Duchenne muscular dystrophy (DMD). Their latest research, published April 12, 2023, in the journal Cell, reveals that in mice, modified viruses, engineered with Myomaker and Myomerger, result in specific fusion with muscle cells. These viruses can therefore be used as a vector to deliver a vital gene needed for muscle function that is mutated in people with DMD.
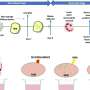
A key unknown prior to this work was whether proteins like Myomaker and Myomerger, which mainly function on cells, could even work on viruses. First author Sajedah Hindi, Ph.D., also with the Division of Molecular Cardiovascular Biology at Cincinnati Children's and a leading member of the research team, took on the challenge to test this idea.
Hindi first designed a strategy to place Myomaker and Myomerger on the surface of viruses and showed that they were functional in cultured cells. She went on to leverage her extensive experience in skeletal muscle biology to test the efficacy of these novel vectors in mice.
"This modified viral vector appears to be a promising tool for delivering a potential lifelong supply of the gene that is absent in people with DMD," Millay says. "The unique advantages of this vector provide an opportunity to significantly impact gene therapy for a myriad of muscle diseases."
What is Duchenne muscular dystrophy?
DMD is a rare and fatal genetic muscle disease characterized by the lack of a critical membrane-stabilizing protein called dystrophin, which results in progressive muscle degeneration and weakness. DMD primarily strikes boys, occurring in about 1 of every 3,500 male births worldwide.
Doctors often diagnose the disease between ages 3 and 6 when children show early signs of significant muscle weakness, such as delayed ability to sit, stand, or walk and difficulties learning to speak. Over time, DMD becomes fatal as muscle degeneration disrupts lung and heart function.
There is no cure. However, lifespans have been extended and quality of life has been improved for many through physical therapy and medications to address certain symptoms. Some gene therapy clinical trials are evaluating the use of adeno-associated virus (AAV) as the delivery vector, and there is hope that these strategies work. However, novel vectors, such as the lentiviruses described by Millay and colleagues, have the potential to improve long-term delivery of therapeutic material for muscular dystrophies.
Modified lentiviruses show benefits in mouse models
Conducting the numerous experiments involved in this study took Hindi and collaborators about four years to complete. A significant collaborator who helped initiate the project was Benjamin Podbilewicz, from the Technion-Israel Institute of Technology, Haifa, Israel. Their findings include:
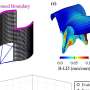
- Demonstrating that the skeletal muscle fusogens Myomaker and Myomerger can replace viral fusogens and fuse viruses to cells.
- Showing specifically that genome-integrating lentiviruses (LV) modified to carry Myomaker (Mymk) and Myomerger (Mymg) can selectively infect skeletal muscle to deliver therapeutic "cargo." In this case, a miniaturized form of the dystrophin protein called "mDystrophin."
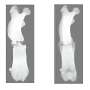
- Documenting that mice mimicking human DMD show improvement after treatment with this approach. Most importantly, the diaphragm muscles got stronger in the treated mice, highlighting the potential to alleviate breathing problems that occur in late-stage DMD.
- Demonstrating that the viral vector reached muscle stem cells, suggesting potential long-lasting benefits from treatment. Improved function lasted more than six months in some treated muscles, while signals of ongoing healthy myofiber production were detected as long as 11 months later.
- Further revealing that the immune systems of treated mice did not recognize the viral vector, suggesting that repeated dosing will be possible if needed.
Next steps
Much more research will be needed to further develop this discovery into a treatment that could someday benefit people with DMD. Even more work will be needed to determine which other muscle diseases might be treated with this lentivirus vector.
"We envision that this concept, transferring a naturally occurring process within muscle to membrane vehicles, could revolutionize delivery of therapeutic material to skeletal muscle to improve genetic conditions such as muscular dystrophy and conditions associated with muscle loss and weakness," Millay says.
More information: Sajedah M. Hindi et al, Enveloped viruses pseudotyped with mammalian myogenic cell fusogens target skeletal muscle for gene delivery, Cell (2023). DOI: 10.1016/j.cell.2023.03.033