Guang Peng, M.D., Ph.D. Credit: MD Anderson Cancer Center
Functional loss of ARID1a, a frequently mutated tumor suppressor gene, causes deficiencies in normal DNA repair and may sensitize tumors to immune checkpoint blockade therapies, according to researchers from The University of Texas MD Anderson Cancer Center. The preclinical study suggests that mutations in ARID1a could be beneficial in predicting immunotherapy success.
The findings, published today in Nature Medicine, are the first to identify a role for ARID1a in regulating mismatch repair (MMR), a normal process for correcting DNA damage. Further, the study showed that treatment with immune checkpoint inhibitors targeting PD-1 successfully reduced tumor burden and prolonged survival in mouse models with ARID1a-deficient tumors relative to controls.
Mutations in ARID1a occur frequently in a wide spectrum of cancers, with particularly high mutation rates (15-50 percent) in clear cell ovarian cancer, endometrial cancer, gastric cancer and bladder cancer. However, most mutations lead to loss of ARID1a, making it a poor therapeutic target, explained senior author Guang Peng, M.D., Ph.D., associate professor of Clinical Cancer Prevention.
"Since this is a very highly mutated gene in cancer, we wanted to better understand the biological function of ARID1a and potential therapeutic vulnerabilities," said Peng. "We did a variety of molecular assays and demonstrated, for the first time, that ARID1a deficiency has a causative relationship with MMR deficiency."
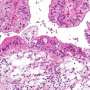
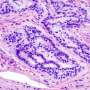
The researchers performed a screen in cancer cells to identify proteins which interact with ARID1a and discovered a connection with MSH2, a protein with a key role in regulating MMR. Additional in vitro assays confirmed that ARID1a was essential to normal MMR function.
Tumors with deficiencies in MMR are known to accumulate large numbers of genetic mutations and corresponding mutant proteins, or neoantigens, as the disease progresses. These neoantigens are thought to stimulate an immune response, making them more susceptible to checkpoint blockade therapy.
The researchers therefore analyzed data across cancer types from The Cancer Genome Atlas (TCGA) and confirmed that tumors with ARID1a mutations indeed carried higher mutational loads. Further, ARID1a mutations were more common in tumors with microsatellite instability (MSI), another marker for MMR dysfunction.
"The FDA has approved MMR deficiency as a marker for the use of checkpoint-blockade immunotherapy," said Peng. "Therefore, we wondered whether ARID1a-deficient tumors would have increased sensitivity to checkpoint blockade because they have impaired MMR and increased mutation load."
Further analysis of TCGA data revealed that tumors with ARID1a mutations exhibited an activation of the immune system, according to gene expression levels of immune markers. Therefore, the researchers investigated the use of checkpoint blockade inhibitors in treating tumors with ARID1a mutations.
Using mouse models of both ovarian and colorectal cancer, the researchers compared the effectiveness of anti-PD-1 therapy in mice with ARID1a-mutant tumors relative to controls with functional ARID1a. Treatment with the checkpoint blockade therapy significantly improved survival in mice with ARID1a mutations, suggesting immunotherapy could be useful for patients with ARID1a-mutant tumors.
"Our findings link ARID1a mutations to MMR deficiency, thus providing a therapeutic target by immune checkpoint blockade," said Peng. "We hope that our data will contribute to clinical studies testing ARID1a as a new biomarker for checkpoint blockade therapies."
Future work will be necessary to confirm the researchers' findings in clinical patient samples. Peng hopes to initiate clinical studies to investigate the value of ARID1a mutations across cancer types as a predictor of response to checkpoint blockade inhibitors targeting PD-1.
Journal information: Nature Medicine
Provided by University of Texas M. D. Anderson Cancer Center