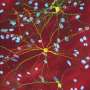
. The neuron in the center (yellow) contains an abnormal intracellular accumulation of huntingtin called an inclusion body (orange). Credit: Wikipedia/ Creative Commons Attribution 3.0 Unported license")
A montage of three images of single striatal neurons transfected with a disease-associated version of huntingtin, the protein that causes Huntington's disease. Nuclei of untransfected neurons are seen in the background (blue). The neuron in the center (yellow) contains an abnormal intracellular accumulation of huntingtin called an inclusion body (orange). Credit: Wikipedia/ Creative Commons Attribution 3.0 Unported license
The region of DNA associated with Huntington's disease has been shown to grow throughout life and contribute towards disease progression.
New research, published in EBioMedicine, reveals that the DNA responsible for Huntington's disease is not stable throughout life, and that older individuals carry longer versions of the genetic mutation than younger individuals.
The researchers also found that individuals whose genetic mutation grew quickest tended to experience an earlier onset of Huntington's disease than expected, and also that their disease progressed more rapidly.
Huntington's disease is a severe, invariably fatal, inherited neurodegenerative brain disorder. It is a rare disease, with only 1 in 10,000 people affected, and is caused by an increase in the number of "CAG' repeats in the DNA of the huntingtin gene. Individuals carrying the critical CAG repeats in their genetic code have a 50% chance of passing the disease to their children. However, in affected families, the number of CAG repeats tends to increase when passed on to children, often resulting in an earlier age at onset of Huntington's disease in succeeding generations.
The study, led by scientists at the University of Glasgow, in association with clinical investigators at University College London and two large teams of international collaborators, investigated the DNA of two large groups of individuals carrying the Huntington's disease mutation.
Researchers were able to show that, as well as the mutation growing over time, the precise sequence of the huntingtin gene was also critical. Specifically, they found that extra "CAA' interruptions in the CAG repeats found in some people slowed down the rate at which the number of CAG repeats grew, and typically resulted in a later age at onset and slower disease progression than expected. They also found that common genetic differences in several DNA repair genes were associated with the rate of CAG growth.
These findings have potential implications for diagnostic testing, which currently only estimates the number of CAG repeats and does not determine the exact DNA sequence.
Professor Darren Monckton, from the University of Glasgow and senior author of the study, said: "These findings offer us further insights into the mechanisms of Huntington's disease—how it develops, what causes it to appear earlier and progress more rapidly.
"Based on our research, treatments aimed at stopping or slowing the rate at which the number of CAG repeats grow could be beneficial. Importantly, the DNA repair genes that have been identified as modifying disease severity, represent novel drug targets for Huntington's disease."
To carry out this research the researchers developed a novel high-throughput DNA sequencing method to examine the dynamics of the CAG repeat in the blood cell DNA of individuals carrying the Huntington's disease mutation.
Most unaffected people in the general population have less than 30 CAG repeats, whereas individuals with Huntington's disease usually have 40 or more CAG repeats. The more CAG repeats a person inherits, the earlier the symptoms of Huntington's disease tend to present.
The paper, "A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington's disease clinical outcomes," is published in EBioMedicine.
More information: A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington's disease clinical outcomes, EBioMedicine.
Journal information: EBioMedicine
Provided by University of Glasgow
.jpg)