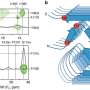
, depicted in blue, binds different DNA aptamers, that have different conformations (hairpin, red; extended, black). Upon binding to these DNA sequences, PrP phase separates, and, depending on the conformation of the nucleic acid, a solid-like structure is formed (green), similar to the one found in the brain of carriers of neurodegenerative diseases related to protein misfolding, such as amyotrophic lateral sclerosis (ALS), and Parkinson's Disease. Credit: Carolina Matos and Anderson Pinheiro")
The globular domain of the prion protein (PrP90-231), depicted in blue, binds different DNA aptamers, that have different conformations (hairpin, red; extended, black). Upon binding to these DNA sequences, PrP phase separates, and, depending on the conformation of the nucleic acid, a solid-like structure is formed (green), similar to the one found in the brain of carriers of neurodegenerative diseases related to protein misfolding, such as amyotrophic lateral sclerosis (ALS), and Parkinson's Disease. Credit: Carolina Matos and Anderson Pinheiro
Researchers at the Federal University of Rio de Janeiro (UFRJ), in Brazil, have identified that the interaction between prion proteins and DNA may be behind the formation of protein amyloid aggregates and of the emergence of neurodegenerative diseases such as Creutzfeldt-Jakob disease and other spongiform encephalopathies. The study appears today in the FASEB Journal.
Led by UFRJ Professors Yraima Cordeiro and Anderson Pinheiro, the scientists have found that the prion protein (PrP) suffers liquid-liquid phase separation, and that this mechanism is finely controlled by some DNA sequences. In a process similar to oil droplets dispersed in an oil-water emulsion, the DNA leads PrP to form liquid droplets, turning it into a gel-like state or even changing them into a solid. They have also observed that these properties depend on the conformation of the DNA aptamer (a hairpin or extended conformation) and on the stoichiometry of the protein-nucleic acid interaction.
The process of turning liquid droplets into a solid state could explain the formation of abnormal and irreversible clumping of the prion protein, known as amyloid aggregates. These structures are toxic to the brain and are related to the development of transmissible spongiform encephalopathies, such as the Creutzfeldt-Jakob disease and the bovine spongiform encephalopathy (BSE), commonly known as mad cow disease. The link between amyloid aggregates and the diseases has been known for years, but how these structures form remains unclear. The study brings insights that might help answer this question.
The main findings of the research enlighten the property of the prion protein to bind nucleic acids in a similar fashion of well-described proteins that cause other neurodegenerative diseases. This opens the possibility of targeting the disease by selecting specific DNA sequences to control or avoid the organelles turning into non-functional gels and solids.
More information: Carolina O. Matos et al, Liquid‐liquid phase separation and fibrillation of the prion protein modulated by a high‐affinity DNA aptamer, The FASEB Journal (2019). DOI: 10.1096/fj.201901897R
Journal information: FASEB Journal
Provided by Instituto Nacional de Ciência e Tecnologia de Biologia Estrutural e Bioimagem (INBEB)